Technische Daten
| Formel | C23H19N3O2 |
||||||
| Molekulargewicht | 369.42 | CAS-Nr. | 905854-02-6 | ||||
| Löslichkeit (25°C)* | In vitro | DMSO | 74 mg/mL (200.31 mM) | ||||
| Ethanol | 40 mg/mL (108.27 mM) | ||||||
| Water | Insoluble | ||||||
| In vivo (Lösungsmittel einzeln und der Reihe nach zum Produkt hinzufügen.) |
|
||||||
|
* <1 mg/ml bedeutet schwer löslich oder unlöslich. * Bitte beachten Sie, dass Selleck die Löslichkeit aller Verbindungen intern testet und die tatsächliche Löslichkeit geringfügig von veröffentlichten Werten abweichen kann. Dies ist normal und ist auf geringfügige Batch-zu-Batch-Variationen zurückzuführen. * Versand bei Raumtemperatur (Stabilitätstests zeigen, dass dieses Produkt ohne Kühlmaßnahmen versendet werden kann.) |
|||||||
Vorbereitung von Stammlösungen
Biologische Aktivität
| Beschreibung | Tivantinib ist der erste nicht-ATP-kompetitive c-Met-Inhibitor mit einem Ki von 0,355 μM in einem zellfreien Assay, geringer Aktivität gegenüber Ron und keiner Hemmung von EGFR, InsR, PDGFRα oder FGFR1/4. Diese Verbindung induziert einen G2/M-Arrest und Apoptose. | ||
|---|---|---|---|
| Ziele |
|
||
| In vitro | Es wurde gezeigt, dass ARQ-197 HGF/c-Met-induzierte zelluläre Reaktionen in vitro verhindert. Diese Verbindung besitzt Antitumoraktivität; sie hemmt die Proliferation von A549-, DBTRG- und NCI-H441-Zellen mit IC50-Werten von 0,38, 0,45, 0,29 μM. Die Behandlung mit diesem Agens führt zu einer Abnahme der Phosphorylierung der MAPK-Signaltransduktionskaskade und zur Verhinderung von Invasion und Migration. Darüber hinaus führt die ektopische Expression von c-Met in NCI-H661, einer Zelllinie ohne endogene Expression von c-Met, dazu, dass diese ein invasives Phänotyp erwirbt, das ebenfalls durch diese Chemikalie unterdrückt wird. Obwohl die Zugabe zunehmender Konzentrationen dieses Inhibitors die Km von ATP nicht signifikant beeinflusst, verringerte die Exposition von c-Met gegenüber 0,5 μM dieser Substanz die Vmax von c-Met um ungefähr das 3-fache. Die Fähigkeit dieses Moleküls, die Vmax zu verringern, ohne die Km von ATP zu beeinflussen, bestätigte, dass es c-Met über einen nicht-ATP-kompetitiven Mechanismus hemmt und daher für seine hohe Kinaseselektivität verantwortlich sein könnte. Es verhindert humanes rekombinantes c-Met mit einer berechneten Hemmkonstante Ki von ungefähr 355 nM. Obwohl die höchste verwendete ATP-Konzentration 200 μM beträgt, wird die Potenz dieser Verbindung gegenüber c-Met durch die Verwendung von ATP-Konzentrationen von bis zu 1 mM nicht reduziert. Es blockiert die c-Met-Phosphorylierung und nachgeschaltete c-Met-Signalwege. Diese Chemikalie unterdrückt die konstitutive und Liganden-vermittelte c-Met-Autophosphorylierung und damit die c-Met-Aktivität, was wiederum zur Hemmung nachgeschalteter c-Met-Effektoren führt. Seine Induktion der Caspase-abhängigen Apoptosis ist in c-Met-exprimierenden menschlichen Krebszellen, einschließlich HT29-, MKN-45- und MDA-MB-231-Zellen, erhöht. |
||
| In vivo | Alle drei mit Tivantinib behandelten Xenograft-Modelle zeigen eine Reduktion des Tumorwachstums: 66 % im HT29-Modell, 45 % im MKN-45-Modell und 79 % im MDA-MB-231-Modell. In diesen Xenograft-Studien werden keine signifikanten Änderungen des Körpergewichts nach oraler Verabreichung dieser Verbindung in einer Dosis von 200 mg/kg beobachtet. Pharmakodynamisch wird die Phosphorylierung von c-Met in menschlichen Kolon-Xenograft-Tumoren (HT29) durch diese Chemikalie stark gehemmt, wie durch eine drastische Reduktion der c-Met-Autophosphorylierung 24 Stunden nach einer einzelnen oralen Dosis von 200 mg/kg dieses Agens festgestellt wurde. Dieselbe Dosierung bei Mäusen zeigt, dass Tumorxenografts anhaltenden Plasmaspiegeln der Verbindung ausgesetzt sind, was mit der beobachteten pharmakodynamischen Hemmung der c-Met-Phosphorylierung und der Hemmung der Proliferation von c-Met-tragenden Krebszelllinien übereinstimmt. Die Plasmaspiegel des Agens 10 Stunden nach der Dosierung werden auf 1,3 μM bestimmt, mehr als das 3-fache über der biochemischen Hemmkonstante dieser Substanz für c-Met. Daher ist es in der Lage, sein Target in vivo im xenograftierten menschlichen Tumorgewebe zu unterdrücken. Zusammenfassend blockiert dieser Inhibitor das Wachstum von c-Met-abhängigen xenograftierten menschlichen Tumoren. |
||
| Merkmale | Der erste selektive c-Met-Inhibitor, der in humane klinische Studien eingebracht wurde. |
Protokoll (aus Referenz)
| Kinase-Assay: |
|
|---|---|
| Zell-Assay: |
|
| Tierstudie: |
|
Referenzen
|
Kundenproduktvalidierung
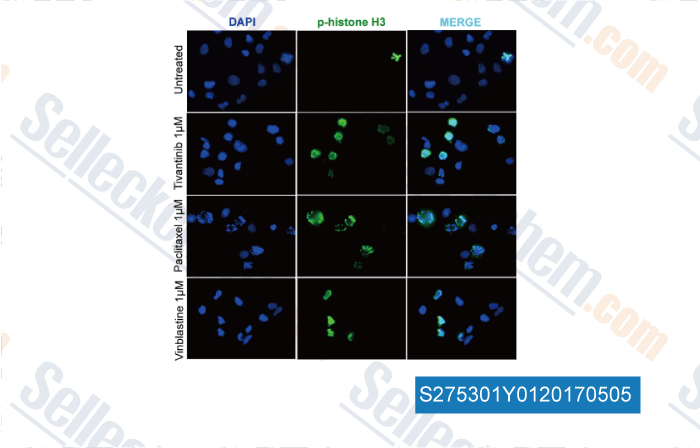
-
, , Clin Cancer Res, 2017, 23(15):4364-4375
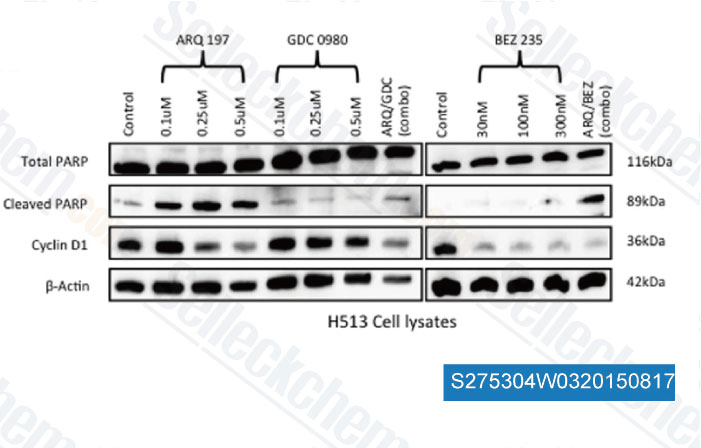
-
Daten von [ , , PLoS One, 2014, 9(9): e105919 ]
Sellecks Tivantinib Wurde zitiert von 50 Publikationen
| MET receptor serves as a promising target in melanoma brain metastases [ Acta Neuropathol, 2024, 147(1):44] | PubMed: 38386085 |
| Insulin receptor substrate 1 is a novel member of EGFR signaling in pancreatic cells [ Eur J Cell Biol, 2024, 103(4):151457] | PubMed: 39326351 |
| EZH2/hSULF1 axis mediates receptor tyrosine kinase signaling to shape cartilage tumor progression [ Elife, 2023, 12e79432] | PubMed: 36622753 |
| A community challenge for a pancancer drug mechanism of action inference from perturbational profile data [ Cell Rep Med, 2022, 3(1):100492] | PubMed: 35106508 |
| High-Resolution Profiling of Lung Adenocarcinoma Identifies Expression Subtypes with Specific Biomarkers and Clinically Relevant Vulnerabilities [ Cancer Res, 2022, 82(21):3917-3931] | PubMed: 36040373 |
| High-resolution profiling of lung adenocarcinoma identifies expression subtypes with specific biomarkers and clinically relevant vulnerabilities [ Cancer Res, 2022, CAN-22-0432] | PubMed: 36040373 |
| Ring Finger Protein 125 Is an Anti-Proliferative Tumor Suppressor in Hepatocellular Carcinoma [ Cancers (Basel), 2022, 14(11)2589] | PubMed: 35681566 |
| Establishment and Characterization of NCC-PMP1-C1: A Novel Patient-Derived Cell Line of Metastatic Pseudomyxoma Peritonei [ J Pers Med, 2022, 12(2)258] | PubMed: 35207746 |
| MTBP enhances the activation of transcription factor ETS-1 and promotes the proliferation of hepatocellular carcinoma cells [ Front Oncol, 2022, 12:985082] | PubMed: 36106099 |
| MET Inhibition Sensitizes Rhabdomyosarcoma Cells to NOTCH Signaling Suppression [ Front Oncol, 2022, 12:835642] | PubMed: 35574376 |
RÜCKGABERICHTLINIE
Die bedingungslose Rückgaberichtlinie von Selleck Chemical gewährleistet unseren Kunden ein reibungsloses Online-Einkaufserlebnis. Wenn Sie in irgendeiner Weise mit Ihrem Kauf unzufrieden sind, können Sie jeden Artikel innerhalb von 7 Tagen nach Erhalt zurückgeben. Im Falle von Produktqualitätsproblemen, sei es protokollbezogene oder produktbezogene Probleme, können Sie jeden Artikel innerhalb von 365 Tagen ab dem ursprünglichen Kaufdatum zurückgeben. Bitte befolgen Sie die nachstehenden Anweisungen, wenn Sie Produkte zurücksenden.
VERSAND UND LAGERUNG
Selleck-Produkte werden bei Raumtemperatur transportiert. Wenn Sie das Produkt bei Raumtemperatur erhalten, seien Sie versichert, dass die Qualitätskontrollabteilung von Selleck Experimente durchgeführt hat, um zu überprüfen, dass die normale Temperaturplatzierung von einem Monat die biologische Aktivität von Pulverprodukten nicht beeinträchtigt. Nach dem Sammeln lagern Sie das Produkt bitte gemäß den in der Datenblatt beschriebenen Anforderungen. Die meisten Selleck-Produkte sind unter den empfohlenen Bedingungen stabil.
NICHT FÜR DIE ANWENDUNG AM MENSCHEN, FÜR VETERINÄRMEDIZINISCHE DIAGNOSTIK ODER THERAPEUTISCHE ZWECKE.