Technische Daten
| Formel | C22H24ClFN4O3 |
||||||
| Molekulargewicht | 446.90 | CAS-Nr. | 184475-35-2 | ||||
| Löslichkeit (25°C)* | In vitro | DMSO | 89 mg/mL (199.14 mM) | ||||
| Ethanol | 4 mg/mL (8.95 mM) | ||||||
| Water | Insoluble | ||||||
| In vivo (Lösungsmittel einzeln und der Reihe nach zum Produkt hinzufügen.) |
|
||||||
|
* <1 mg/ml bedeutet schwer löslich oder unlöslich. * Bitte beachten Sie, dass Selleck die Löslichkeit aller Verbindungen intern testet und die tatsächliche Löslichkeit geringfügig von veröffentlichten Werten abweichen kann. Dies ist normal und ist auf geringfügige Batch-zu-Batch-Variationen zurückzuführen. * Versand bei Raumtemperatur (Stabilitätstests zeigen, dass dieses Produkt ohne Kühlmaßnahmen versendet werden kann.) |
|||||||
Vorbereitung von Stammlösungen
Biologische Aktivität
| Beschreibung | Gefitinib ist ein EGFR-Inhibitor für Tyr1173, Tyr992, Tyr1173 und Tyr992 in den NR6wtEGFR- und NR6W-Zellen mit einer IC50 von 37 nM, 37 nM, 26 nM bzw. 57 nM. Diese Verbindung fördert Autophagy und Apoptosis von Lungenkrebszellen über die Blockade des PI3K/AKT/mTOR-Signalwegs. | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ziele |
|
||||||||
| In vitro | Gefitinib hemmt effektiv alle Tyrosinphosphorylierungsstellen an EGFR in sowohl hoch- als auch niedrig-EGFR-exprimierenden Zelllinien, einschließlich der NR6-, NR6M- und NR6W-Zelllinien. Die Phosphorylierungsstellen Tyr1173 und Tyr992 sind weniger empfindlich und erfordern höhere Konzentrationen dieser Verbindung zur Hemmung. Es blockiert effektiv die Phosphorylierung von PLC-γ mit einer IC50 von 27 nM in NR6W-Zellen. Die NR6wtEGFR- und NR6M-Zelllinien weisen niedrige PLC-γ-Phosphorylierungsspiegel auf, aber der Spiegel in der NR6M-Zelllinie ist resistenter gegenüber der Hemmung durch diese Chemikalie mit einer IC50 von 43 nM bzw. 369 nM. Diese Verbindung hemmt Akt-Phosphorylierungen mit einer IC50 von 220 und 263 nM in den niedrig-EGFR- bzw. -EGFRvIII-exprimierenden Zelllinien. Es fördert im Dosisbereich von 0,1 bis 0,5 μM signifikant, anstatt abzuschwächen, die Koloniebildung von NR6M-Zellen. Bei einer Konzentration von 2 μM blockiert diese Chemikalie jedoch vollständig die NR6M-Koloniebildung. Es hemmt schnell und dosisabhängig die EGFR- und ERK-Phosphorylierung bis zu 72 Stunden nach EGF-Stimulation in sowohl den hoch- als auch niedrig-EGFR-exprimierenden Zelllinien. Diese Verbindung ist das Monolayer-Wachstum dieser EGF-gesteuerten untransformierten MCF10A-Zellen mit einer IC50 von 20 nM. Die Kombination dieser Chemikalie (0,2 μM und 0,5 μM) mit Bestrahlung führte zu einer signifikanten Wachstumshemmung in LoVo-Zellen im Vergleich zur alleinigen Bestrahlung. |
||||||||
| In vivo | Gefitinib (100 mg/kg) verbessert die Antitumorwirkung der Strahlentherapie bei LoVo-Tumor-Xenotransplantaten. Die Behandlung von Nacktmäusen mit etablierten menschlichen GEO-Kolonkarzinom-Xenotransplantaten mit dieser Verbindung zeigt eine reversible dosisabhängige Hemmung des Tumorwachstums, da GEO-Tumoren am Ende der Behandlung die Wachstumsrate der Kontrollen wieder aufnehmen. |
||||||||
| Merkmale | Ein potenter EGFR tyrosine kinase inhibitor. |
Protokoll (aus Referenz)
| Zell-Assay: |
|
|---|---|
| Tierstudie: |
|
Referenzen
|
Kundenproduktvalidierung
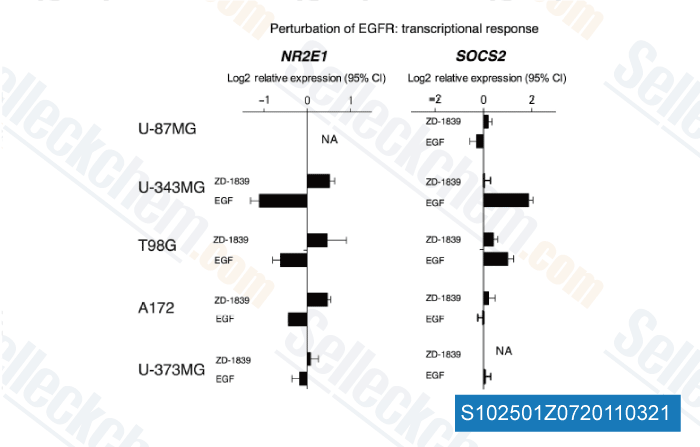
-
Daten von [ Mol Syst Biol , 2011 , 7, 486 ]
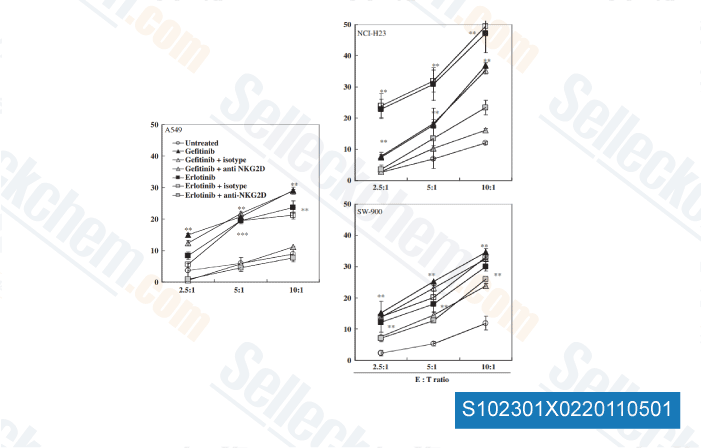
-
Daten von [ J Immunother , 2011 , 34(4), 372-81 ]
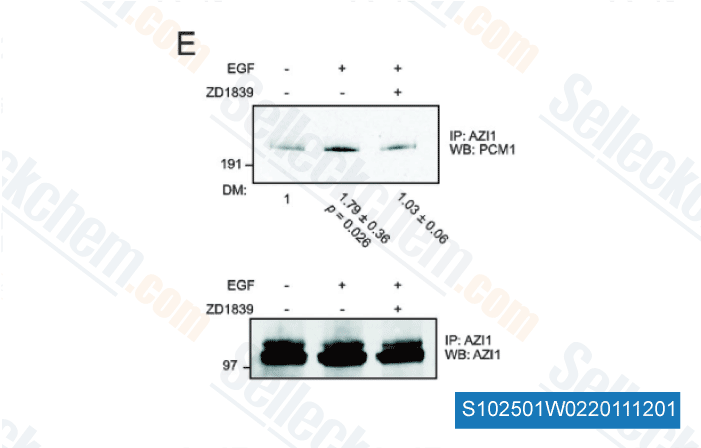
-
Daten von [ Mol Biosyst , 2011 , 7, 3223-33 ]
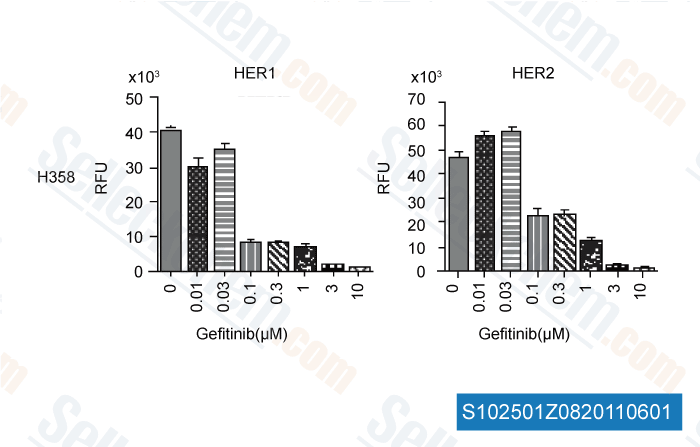
-
Daten von [ Int J Proteomics , 2011 , 215496 ]
Sellecks Gefitinib (ZD1839) Wurde zitiert von 789 Publikationen
| Galectin-3-integrin α5β1 phase separation disrupted by advanced glycation end-products impairs diabetic wound healing in rodents [ Nat Commun, 2025, 16(1):7287] | PubMed: 40775187 |
| Androgen receptor promotes arachidonic acid metabolism and angiogenic microenvironment in AFP-negative hepatocellular carcinoma [ Nat Commun, 2025, 16(1):6451] | PubMed: 40651942 |
| The potential of lazertinib and amivantamab combination therapy as a treatment strategy for uncommon EGFR-mutated NSCLC [ Cell Rep Med, 2025, 6(2):101929] | PubMed: 39874964 |
| Anti-galectin-9 therapy synergizes with EGFR inhibition to reprogram the tumor microenvironment and overcome immune evasion [ J Immunother Cancer, 2025, 13(7)e010926] | PubMed: 40664443 |
| Separase Inhibition Enhances Gefitinib Sensitivity of Lung Cancer via PTBP1/TAK1/RIPK1-Mediated PANoptosis [ MedComm (2020), 2025, 6(11):e70432] | PubMed: 41122447 |
| METTL1/WDR4-mediated m7G Hypermethylation of SCLT1 mRNA Promotes Gefitinib Resistance in NSCLC [ Genomics Proteomics Bioinformatics, 2025, qzaf076] | PubMed: 40857569 |
| N6-methyladenosine-modified GPX2 impacts cancer cell stemness and TKI resistance through regulating of redox metabolism [ Cell Death Dis, 2025, 16(1):458] | PubMed: 40533443 |
| Antitumor activity of afatinib in EGFR T790M-negative human oral cancer therapeutically targets mTOR/Mcl-1 signaling axis [ Cell Oncol (Dordr), 2025, 48(1):123-138] | PubMed: 38888847 |
| Efficacy of Conventional and Novel Tyrosine Kinase Inhibitors for Uncommon EGFR Mutations-An In Vitro Study [ Cells, 2025, 14(17)1386] | PubMed: 40940797 |
| Glimepiride overcomes acquired resistance to EGFR TKIs in lung cancer via the AMPK/ERK/MMP7 signaling pathway [ Biochem Pharmacol, 2025, 241:117162] | PubMed: 40659128 |
RÜCKGABERICHTLINIE
Die bedingungslose Rückgaberichtlinie von Selleck Chemical gewährleistet unseren Kunden ein reibungsloses Online-Einkaufserlebnis. Wenn Sie in irgendeiner Weise mit Ihrem Kauf unzufrieden sind, können Sie jeden Artikel innerhalb von 7 Tagen nach Erhalt zurückgeben. Im Falle von Produktqualitätsproblemen, sei es protokollbezogene oder produktbezogene Probleme, können Sie jeden Artikel innerhalb von 365 Tagen ab dem ursprünglichen Kaufdatum zurückgeben. Bitte befolgen Sie die nachstehenden Anweisungen, wenn Sie Produkte zurücksenden.
VERSAND UND LAGERUNG
Selleck-Produkte werden bei Raumtemperatur transportiert. Wenn Sie das Produkt bei Raumtemperatur erhalten, seien Sie versichert, dass die Qualitätskontrollabteilung von Selleck Experimente durchgeführt hat, um zu überprüfen, dass die normale Temperaturplatzierung von einem Monat die biologische Aktivität von Pulverprodukten nicht beeinträchtigt. Nach dem Sammeln lagern Sie das Produkt bitte gemäß den in der Datenblatt beschriebenen Anforderungen. Die meisten Selleck-Produkte sind unter den empfohlenen Bedingungen stabil.
NICHT FÜR DIE ANWENDUNG AM MENSCHEN, FÜR VETERINÄRMEDIZINISCHE DIAGNOSTIK ODER THERAPEUTISCHE ZWECKE.