nur für Forschungszwecke
Abacavir Reverse Transcriptase Inhibitor
Kat.-Nr.S5215
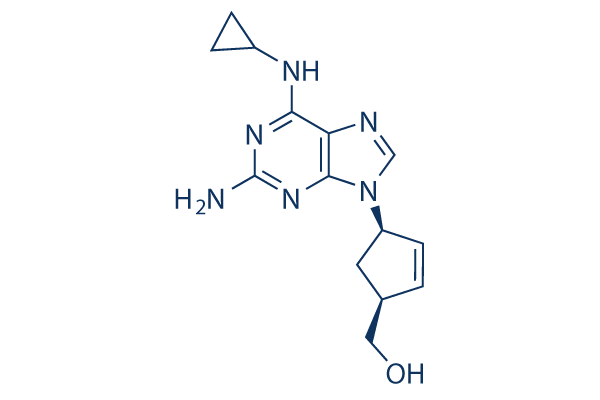
Chemische Struktur
Molekulargewicht: 286.33
Qualitätskontrolle
| Verwandte Ziele | Integrase Bacterial Antibiotics Anti-infection Fungal Antiviral COVID-19 Parasite HIV HCV Protease |
|---|---|
| Weitere Reverse Transcriptase Inhibitoren | Dapivirine (TMC120) Fangchinoline Salicylanilide 3'-Fluoro-3'-deoxythymidine (Alovudine) Ulonivirine Lersivirine (UK-453061) Bifendate 4-Chloro-2-(trifluoroacetyl)aniline hydrochloride |
Chemische Informationen, Lagerung & Stabilität
| Molekulargewicht | 286.33 | Formel | C14H18N6O |
Lagerung (Ab dem Eingangsdatum) | |
|---|---|---|---|---|---|
| CAS-Nr. | 136470-78-5 | -- | Lagerung von Stammlösungen |
|
|
| Synonyme | 1592U89 | Smiles | C1CC1NC2=C3C(=NC(=N2)N)N(C=N3)C4CC(C=C4)CO | ||
Löslichkeit
|
In vitro |
DMSO
: 57 mg/mL
(199.07 mM)
Ethanol : 57 mg/mL Water : Insoluble |
Molaritätsrechner
|
In vivo |
|||||
In-vivo-Formulierungsrechner (Klare Lösung)
Schritt 1: Geben Sie die untenstehenden Informationen ein (Empfohlen: Ein zusätzliches Tier zur Berücksichtigung von Verlusten während des Experiments)
Schritt 2: Geben Sie die In-vivo-Formulierung ein (Dies ist nur der Rechner, keine Formulierung. Bitte kontaktieren Sie uns zuerst, wenn es im Abschnitt "Löslichkeit" keine In-vivo-Formulierung gibt.)
Berechnungsergebnisse:
Arbeitskonzentration: mg/ml;
Methode zur Herstellung der DMSO-Stammlösung: mg Wirkstoff vorgelöst in μL DMSO ( Konzentration der Stammlösung mg/mL, Bitte kontaktieren Sie uns zuerst, wenn die Konzentration die DMSO-Löslichkeit der Wirkstoffcharge überschreitet. )
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügenμL PEG300, mischen und klären, dann hinzufügenμL Tween 80, mischen und klären, dann hinzufügen μL ddH2O, mischen und klären.
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügen μL Maisöl, mischen und klären.
Hinweis: 1. Bitte stellen Sie sicher, dass die Flüssigkeit klar ist, bevor Sie das nächste Lösungsmittel hinzufügen.
2. Achten Sie darauf, das/die Lösungsmittel der Reihe nach hinzuzufügen. Sie müssen sicherstellen, dass die bei der vorherigen Zugabe erhaltene Lösung eine klare Lösung ist, bevor Sie mit der Zugabe des nächsten Lösungsmittels fortfahren. Physikalische Methoden wie Vortex, Ultraschall oder ein heißes Wasserbad können zur Unterstützung des Lösens verwendet werden.
Wirkmechanismus
| Targets/IC50/Ki |
Reverse transcriptase
|
|---|---|
| In vitro |
Abacavir (ABC) zeigt eine potente antivirale Aktivität in vitro gegen Wildtyp-HIV-1 (IC50 4,0 μM, MT-4-Zellen). Diese Verbindung induziert chromosomale DSBs und tötet dadurch ATL-Zellen, aber keine nicht mit HTLV-1 infizierten Zellen. Nach der Aufnahme in die Zellen wird es in einem einzigartigen schrittweisen Anabolismus phosphoryliert, um in das triphosphatierte Guanin-Analogon Carbovir (CBV) umgewandelt und dann von replikativen DNA-Polymerasen in die chromosomale Wirts-DNA eingebaut zu werden, was zu einem vorzeitigen Abbruch der DNA-Replikation, dem Kollaps der Replikationsgabel und der Bildung von DSBs führt. Diese Chemikalie induziert einen S/G2-Phasen-Arrest und Apoptose in ED-40515(−)-Zellen, jedoch nicht in Jurkat-Zellen.
|
| In vivo |
Abacavir hemmt effizient das Wachstum von ATL-Zell-Xenografts in NOD/SCID-Mäusen. Bei Erwachsenen wird diese Verbindung nach oraler Verabreichung schnell resorbiert, wobei Spitzenkonzentrationen 0,63-1 Stunde nach der Dosis auftreten. Die absolute Bioverfügbarkeit von Abacavir beträgt etwa 83%. Seine Pharmakokinetik ist linear und dosisproportional im Bereich von 300-1200 mg/Tag. Das scheinbare Verteilungsvolumen nach intravenöser Verabreichung beträgt etwa 0,86 ± 0,15 L/kg, was darauf hindeutet, dass es in extravaskuläre Räume verteilt wird. Die Bindung an Plasmaproteine beträgt etwa 50% und ist unabhängig von der Plasma-Abacavir-Konzentration. Diese Chemikalie wird in der Leber extensiv metabolisiert; weniger als 2% werden als unverändertes Medikament im Urin ausgeschieden. Es wird hauptsächlich über zwei Wege metabolisiert, Uridindiphosphat-Glucuronyltransferase und Alkoholdehydrogenase, was zum inaktiven Glucuronid-Metaboliten und dem inaktiven Carboxylat-Metaboliten führt. Die terminale Eliminationshalbwertszeit beträgt etwa 1,5 Stunden. Der antivirale Effekt ist auf seinen intrazellulären Anaboliten, Carbovirtriphosphat (CBV-TP), zurückzuführen. Diese Verbindung wird nicht signifikant von Cytochrom P450 (CYP)-Enzymen metabolisiert, noch hemmt sie diese Enzyme.
|
Literatur |
|
Klinische Studieninformationen
(Daten von https://clinicaltrials.gov, aktualisiert am 2024-05-22)
| NCT-Nummer | Rekrutierung | Erkrankungen | Sponsor/Kooperationspartner | Startdatum | Phasen |
|---|---|---|---|---|---|
| NCT04133012 | Completed | HIV-1 Infection |
ANRS Emerging Infectious Diseases|ViiV Healthcare |
February 10 2020 | Not Applicable |
| NCT02708342 | Completed | HIV |
IRCCS San Raffaele|Merck Sharp & Dohme LLC |
April 2016 | -- |
Technischer Support
Tel: +1-832-582-8158 Ext:3
Wenn Sie weitere Fragen haben, hinterlassen Sie bitte eine Nachricht.
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.