nur für Forschungszwecke
AZ876 Liver X Receptor Agonist
Kat.-Nr.S6427
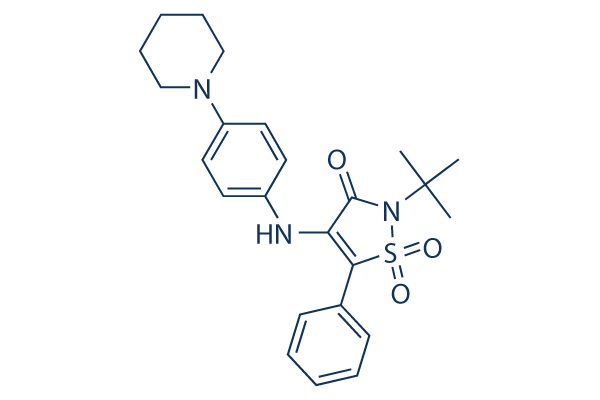
Chemische Struktur
Molekulargewicht: 439.57
Qualitätskontrolle
- In Nature Medicine für seine erstklassige Qualität zitiert
- COA
- NMR
- HPLC
- SDS
- Datenblatt
| Verwandte Ziele | Dehydrogenase HSP Transferase P450 (e.g. CYP17) PDE phosphatase PPAR Vitamin Carbohydrate Metabolism Mitochondrial Metabolism |
|---|---|
| Weitere Liver X Receptor Inhibitoren | GW3965 Hydrochloride T0901317 LXR-623 (WAY-252623) GSK2033 SR9243 Abequolixron (RGX-104) GSK3987 Desmosterol |
Chemische Informationen, Lagerung & Stabilität
| Molekulargewicht | 439.57 | Formel | C24H29N3O3S |
Lagerung (Ab dem Eingangsdatum) | 3 years -20°C powder |
|---|---|---|---|---|---|
| CAS-Nr. | 898800-26-5 | -- | Lagerung von Stammlösungen |
|
|
| Synonyme | N/A | Smiles | CC(C)(C)N1C(=O)C(=C(S1(=O)=O)C2=CC=CC=C2)NC3=CC=C(C=C3)N4CCCCC4 | ||
Löslichkeit
|
In vitro |
DMSO
: 88 mg/mL
(200.19 mM)
|
Molaritätsrechner
|
In vivo |
|||||
In-vivo-Formulierungsrechner (Klare Lösung)
Schritt 1: Geben Sie die untenstehenden Informationen ein (Empfohlen: Ein zusätzliches Tier zur Berücksichtigung von Verlusten während des Experiments)
Schritt 2: Geben Sie die In-vivo-Formulierung ein (Dies ist nur der Rechner, keine Formulierung. Bitte kontaktieren Sie uns zuerst, wenn es im Abschnitt "Löslichkeit" keine In-vivo-Formulierung gibt.)
Berechnungsergebnisse:
Arbeitskonzentration: mg/ml;
Methode zur Herstellung der DMSO-Stammlösung: mg Wirkstoff vorgelöst in μL DMSO ( Konzentration der Stammlösung mg/mL, Bitte kontaktieren Sie uns zuerst, wenn die Konzentration die DMSO-Löslichkeit der Wirkstoffcharge überschreitet. )
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügenμL PEG300, mischen und klären, dann hinzufügenμL Tween 80, mischen und klären, dann hinzufügen μL ddH2O, mischen und klären.
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügen μL Maisöl, mischen und klären.
Hinweis: 1. Bitte stellen Sie sicher, dass die Flüssigkeit klar ist, bevor Sie das nächste Lösungsmittel hinzufügen.
2. Achten Sie darauf, das/die Lösungsmittel der Reihe nach hinzuzufügen. Sie müssen sicherstellen, dass die bei der vorherigen Zugabe erhaltene Lösung eine klare Lösung ist, bevor Sie mit der Zugabe des nächsten Lösungsmittels fortfahren. Physikalische Methoden wie Vortex, Ultraschall oder ein heißes Wasserbad können zur Unterstützung des Lösens verwendet werden.
Wirkmechanismus
| Targets/IC50/Ki |
LXRα
0.007 μM(Ki)
LXRβ
0.011 μM(Ki)
|
|---|---|
| In vitro |
In Bindungsstudien ist AZ876 25-fach und 2,5-fach potenter als GW3965 auf menschliches (h)LXRα bzw. hLXRβ. In Reporter-Transaktivierungsstudien ist diese Verbindung 196-fach und fünffach potenter als GW3965 auf hLXRα bzw. hLXRβ. Sie ist auch potenter als GW3965 auf Maus (m)LXRα (248-fach) und mLXRβ (10,5-fach). Diese Chemikalie ist vier- bis siebenfach potenter als GW3965 auf die Expression von ABCA1-mRNA in Hamster- und menschlichen Blut-PMN-Zellen. Sie ist hochselektiv in Bezug auf andere nukleäre Hormonrezeptoren, einschließlich Retinoid X Rezeptor, Farnesoid X Rezeptor, Thyroid hormone receptor (TR)α oder TRβ, wenn sie im Agonistenmodus in Fluoreszenzresonanzenergietransfer-Assays getestet wurde. |
| In vivo |
AZ876 ist ein dualer partieller LXR-Agonist, der in niedriger Dosis verabreicht bei Mäusen die Atherosklerose reduziert, ohne die Leber- oder Plasma-Triglyceridspiegel zu beeinflussen. Die chronische Verabreichung dieser Verbindung mildert die pathologische kardiale Hypertrophie in einem Mausmodell der chronischen Drucküberlastung, ohne den systemischen Blutdruck zu verändern, was auf herzspezifische Effekte hindeutet. Diese Verbindungstherapie verringert die Myokardfibrose und unterdrückt die Induktion der profibrotischen Genexpression. |
Literatur |
|
Technischer Support
Tel: +1-832-582-8158 Ext:3
Wenn Sie weitere Fragen haben, hinterlassen Sie bitte eine Nachricht.
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.