nur für Forschungszwecke
GSK1838705A ALK Inhibitor
Kat.-Nr.S2703
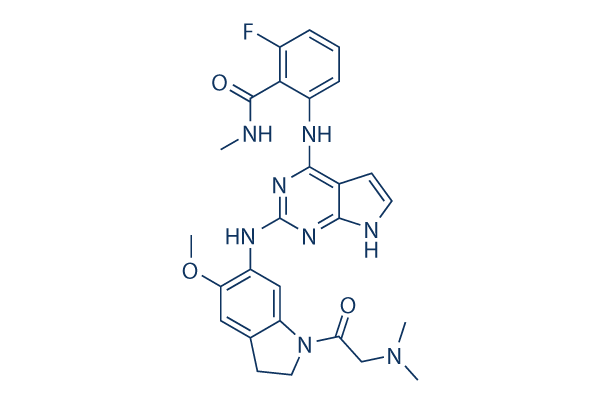
Chemische Struktur
Molekulargewicht: 532.57
Qualitätskontrolle
| Verwandte Ziele | EGFR VEGFR PDGFR FGFR c-Met Src MEK CSF-1R FLT3 HER2 |
|---|---|
| Weitere ALK Inhibitoren | TAE684 (NVP-TAE684) Repotrectinib (TPX-0005) AZD3463 Ensartinib dihydrochloride AP26113-analog (ALK-IN-1) ASP3026 NVL-655 (Neladalkib) Envonalkib Belizatinib (TSR-011) HG-14-10-04 |
Chemische Informationen, Lagerung & Stabilität
| Molekulargewicht | 532.57 | Formel | C27H29FN8O3 |
Lagerung (Ab dem Eingangsdatum) | |
|---|---|---|---|---|---|
| CAS-Nr. | 1116235-97-2 | SDF herunterladen | Lagerung von Stammlösungen |
|
|
| Synonyme | N/A | Smiles | CNC(=O)C1=C(C=CC=C1F)NC2=NC(=NC3=C2C=CN3)NC4=C(C=C5CCN(C5=C4)C(=O)CN(C)C)OC | ||
Löslichkeit
|
In vitro |
DMSO
: 107 mg/mL
(200.91 mM)
Water : Insoluble Ethanol : Insoluble |
Molaritätsrechner
|
In vivo |
|||||
In-vivo-Formulierungsrechner (Klare Lösung)
Schritt 1: Geben Sie die untenstehenden Informationen ein (Empfohlen: Ein zusätzliches Tier zur Berücksichtigung von Verlusten während des Experiments)
Schritt 2: Geben Sie die In-vivo-Formulierung ein (Dies ist nur der Rechner, keine Formulierung. Bitte kontaktieren Sie uns zuerst, wenn es im Abschnitt "Löslichkeit" keine In-vivo-Formulierung gibt.)
Berechnungsergebnisse:
Arbeitskonzentration: mg/ml;
Methode zur Herstellung der DMSO-Stammlösung: mg Wirkstoff vorgelöst in μL DMSO ( Konzentration der Stammlösung mg/mL, Bitte kontaktieren Sie uns zuerst, wenn die Konzentration die DMSO-Löslichkeit der Wirkstoffcharge überschreitet. )
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügenμL PEG300, mischen und klären, dann hinzufügenμL Tween 80, mischen und klären, dann hinzufügen μL ddH2O, mischen und klären.
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügen μL Maisöl, mischen und klären.
Hinweis: 1. Bitte stellen Sie sicher, dass die Flüssigkeit klar ist, bevor Sie das nächste Lösungsmittel hinzufügen.
2. Achten Sie darauf, das/die Lösungsmittel der Reihe nach hinzuzufügen. Sie müssen sicherstellen, dass die bei der vorherigen Zugabe erhaltene Lösung eine klare Lösung ist, bevor Sie mit der Zugabe des nächsten Lösungsmittels fortfahren. Physikalische Methoden wie Vortex, Ultraschall oder ein heißes Wasserbad können zur Unterstützung des Lösens verwendet werden.
Wirkmechanismus
| Merkmale |
A small-molecule kinase inhibitor of IGF-1R and the insulin receptor.
|
|---|---|
| Targets/IC50/Ki |
ALK
(Cell-free assay) 0.5 nM
Insulin Receptor
(Cell-free assay) 1.6 nM
IGF-1R
(Cell-free assay) 2 nM
|
| In vitro |
GSK1838705A hemmt potent und ATP-kompetitiv IGF-1R und IR mit appKi-Werten von 0,7 nM bzw. 1,1 nM. In Zellen hemmt diese Verbindung potent die Liganden-induzierte Phosphorylierung von IGF-1R und IR mit einer IC50 von 85 nM bzw. 79 nM. Sie zeigt eine signifikante antiproliferative Wirkung in einer Reihe von Zelllinien, die aus soliden und hämatologischen Tumoren wie L-82-, SUP-M2-, SK-ES- und MCF-7-Zellen stammen, mit einer EC50 von 24 nM, 28 nM, 141 nM bzw. 203 nM. Diese Chemikalie zeigt eine Akkumulation von MCF-7- und NCl-H929-Zellen überwiegend in der G1 (2N)-Phase des Zellzyklus. Sie hemmt auch ALK mit einem Ki von 0,35 nM und unterdrückt die Proliferation von Nukleophosmin (NPM)-ALK-Fusionszellen mit einer EC50 von 24-88 nM. Diese Verbindung hemmt potent die NPM-ALK-Phosphorylierung in Karpas-299- und SR-786-Zellen, während sie eine moderate Wirkung auf die STAT3-Phosphorylierung hat.
|
| Kinase-Assay |
Kinase-Assays
|
|
Baculovirus-exprimierte Glutathion-S-Transferase-markierte Proteine, die die intrazelluläre Domäne von IGF-1R (Aminosäuren 957-1367) und IR (Aminosäuren 979-1382) kodieren, werden zur Bestimmung der IC50s mittels eines homogenen zeitaufgelösten Fluoreszenzassays verwendet. Ein Filterbindungsassay wird zur Bestimmung der appKi unter Verwendung aktivierter IGF-1R- und IR-Kinasen verwendet. Ein erweitertes Kinase-Selektivitätsprofil dieser Verbindung wird durch Screening im KinaseProfiler-Panel durchgeführt.
|
|
| In vivo |
Bei NIH-3T3/LISN-Tumor-tragenden Mäusen führt die orale Behandlung mit GSK1838705A (60 mg/kg) zu einer Tumorwachstumshemmung um 77 %, ohne signifikanten Gewichtsverlust. Bei COLO 205-Tumor-tragenden Mäusen beträgt die Hemmung des Tumorwachstums durch diese Verbindung (30 mg/kg) 80 %. Darüber hinaus wird die Antitumorwirksamkeit dieser Chemikalie auch bei Mäusen mit HT29-Xenograft oder BxPC3-Xenograft beobachtet. Bei Mäusen führt diese Verbindung (60 mg/kg) zu einem vorübergehenden 2-fachen Anstieg des Blutzuckerspiegels durch Hemmung der IR-Signalgebung. Sie (60 mg/kg) hemmt das Wachstum etablierter Karpas-299-Xenografts mit 93 % Tumorwachstumshemmung, ohne Auswirkungen auf das Gewicht der Ratten.
|
Literatur |
Technischer Support
Tel: +1-832-582-8158 Ext:3
Wenn Sie weitere Fragen haben, hinterlassen Sie bitte eine Nachricht.
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.