nur für Forschungszwecke
Ipragliflozin (ASP1941) SGLT Inhibitor
Kat.-Nr.S8637
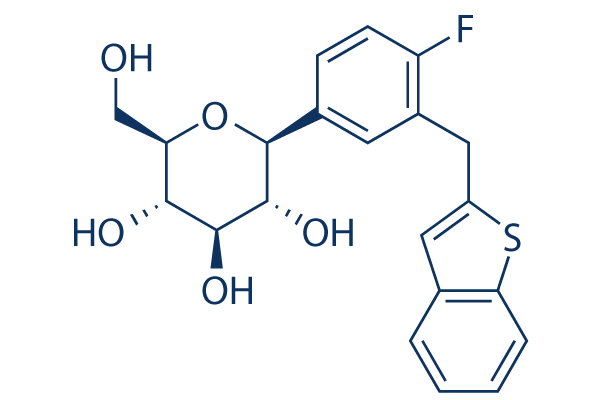
Chemische Struktur
Molekulargewicht: 404.45
Qualitätskontrolle
- In Nature Medicine für seine erstklassige Qualität zitiert
- COA
- NMR
- SDS
- Datenblatt
Chemische Informationen, Lagerung & Stabilität
| Molekulargewicht | 404.45 | Formel | C21H21FOS |
Lagerung (Ab dem Eingangsdatum) | |
|---|---|---|---|---|---|
| CAS-Nr. | 761423-87-4 | SDF herunterladen | Lagerung von Stammlösungen |
|
|
| Synonyme | N/A | Smiles | C1=CC=C2C(=C1)C=C(S2)CC3=C(C=CC(=C3)C4C(C(C(C(O4)CO)O)O)O)F | ||
Löslichkeit
|
In vitro |
DMSO
: 80 mg/mL
(197.79 mM)
Ethanol : 80 mg/mL Water : Insoluble |
Molaritätsrechner
|
In vivo |
|||||
In-vivo-Formulierungsrechner (Klare Lösung)
Schritt 1: Geben Sie die untenstehenden Informationen ein (Empfohlen: Ein zusätzliches Tier zur Berücksichtigung von Verlusten während des Experiments)
Schritt 2: Geben Sie die In-vivo-Formulierung ein (Dies ist nur der Rechner, keine Formulierung. Bitte kontaktieren Sie uns zuerst, wenn es im Abschnitt "Löslichkeit" keine In-vivo-Formulierung gibt.)
Berechnungsergebnisse:
Arbeitskonzentration: mg/ml;
Methode zur Herstellung der DMSO-Stammlösung: mg Wirkstoff vorgelöst in μL DMSO ( Konzentration der Stammlösung mg/mL, Bitte kontaktieren Sie uns zuerst, wenn die Konzentration die DMSO-Löslichkeit der Wirkstoffcharge überschreitet. )
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügenμL PEG300, mischen und klären, dann hinzufügenμL Tween 80, mischen und klären, dann hinzufügen μL ddH2O, mischen und klären.
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügen μL Maisöl, mischen und klären.
Hinweis: 1. Bitte stellen Sie sicher, dass die Flüssigkeit klar ist, bevor Sie das nächste Lösungsmittel hinzufügen.
2. Achten Sie darauf, das/die Lösungsmittel der Reihe nach hinzuzufügen. Sie müssen sicherstellen, dass die bei der vorherigen Zugabe erhaltene Lösung eine klare Lösung ist, bevor Sie mit der Zugabe des nächsten Lösungsmittels fortfahren. Physikalische Methoden wie Vortex, Ultraschall oder ein heißes Wasserbad können zur Unterstützung des Lösens verwendet werden.
Wirkmechanismus
| Targets/IC50/Ki |
mouse SGLT2
(cell-free) 5.64 nM
rat SGLT2
(cell-free) 6.73 nM
hSGLT2
(cell-free) 7.4 nM
|
|---|---|
| In vitro |
Ipragliflozin (ASP1941) hemmt konzentrationsabhängig die SGLT2-Aktivität bei Mäusen, Ratten und Menschen in nanomolaren Konzentrationen. Darüber hinaus hemmt es nicht potent die menschlichen SGLT4- und SGLT5-Isoformen (IC50>1.000 nM). Außerdem hemmt diese Verbindung verschiedene Glukosetransporter (GLUT)-Isoformen, einschließlich GLUT1 und GLUT4, in Maus-3T3-L1-, Ratten-L6-, menschlichen Caco-2- und HepG2-Zellen nicht (IC50>1.000 nM). Es interagiert nicht mit verschiedenen Rezeptoren, Ionenkanälen und Transportern wie adrenergen (α1, α2 und β), muskarinischen (M1, M2 und nicht-selektiv), Angiotensin (AT1 und AT2), Calciumkanal (L-Typ und N-Typ), Kaliumkanal (KATP und SKCa), Natriumkanal (Stelle 2), Cholezystokinin (CCKA und CCKB), Dopamin (D1, D2 und Transporter), Endothelin (ETA und ETB), Gamma-Aminobuttersäure (GABAA und GABAB), Glutamat (AMPA, Kainat und NMDA), Serotonin (5-HT1, 5HT2B und Transporter), Histamin (H1, H2 und H3) und Neurokinin (NK1, NK2 und NK3), wobei die IC50-Werte >3.000 nM betragen. Es ist stabil gegenüber intestinalen Glukosidasen von Mäusen.
|
| In vivo |
Einzelne orale Dosen (0,01-10 mg/kg) von Ipragliflozin (ASP1941) induzieren eine dosisabhängige Harnzuckerausscheidung sowohl bei normalen als auch bei KK-Ay-Mäusen, einem Typ-2-Diabetes-Modell. Einzelne Verabreichungen dieser Verbindung (0,1, 0,3 und 1 mg/kg) reduzieren dosisabhängig den Blutzuckerspiegel sowohl bei KK-Ay-Mäusen als auch bei STZ-Ratten. Die Verabreichung einer einzelnen Dosis von 0,3 mg/kg intravenös und einer einzelnen Dosis von 1 mg/kg oral an Ratten zeigt, dass es eine gute Bioverfügbarkeit mit einem Wert von 71,7% aufweist. Es zeigt gute pharmakokinetische Eigenschaften nach oraler Verabreichung und erhöht dosisabhängig die Harnzuckerausscheidung, die bei normalen Mäusen über 12 Stunden anhält. Die Einzelverabreichung führt zu dosisabhängigen und anhaltenden antihyperglykämischen Effekten in beiden Diabetesmodellen. Es hat ein geringes Hypoglykämie-Risiko. Nach oraler Verabreichung (3 mg/kg) an normale Mäuse erreichen die Plasmakonzentrationen nach 1 Stunde ein Maximum und nehmen dann allmählich ab. Selbst 8 Stunden nach der Verabreichung werden deutliche Plasmakonzentrationen nachgewiesen. In den pharmakokinetischen Studien an Mäusen zeigt es eine gute orale Bioverfügbarkeit und weist über lange Zeiträume hohe Arzneimittelkonzentrationen auf. Die absolute Bioverfügbarkeit beträgt 71,7-90,7% bei Ratten und 74,5-75,3% bei Affen.
|
Literatur |
|
Klinische Studieninformationen
(Daten von https://clinicaltrials.gov, aktualisiert am 2024-05-22)
| NCT-Nummer | Rekrutierung | Erkrankungen | Sponsor/Kooperationspartner | Startdatum | Phasen |
|---|---|---|---|---|---|
| NCT02794792 | Completed | Type 2 Diabetes Mellitus |
Astellas Pharma Europe B.V.|Astellas Pharma Inc |
May 11 2016 | Phase 3 |
| NCT02529449 | Completed | Type 1 Diabetes Mellitus |
Astellas Pharma Inc |
September 1 2015 | Phase 2 |
| NCT01972880 | Completed | Healthy|Plasma Concentration of ASP1941 |
Astellas Pharma Inc |
September 2013 | Phase 1 |
| NCT01611363 | Completed | Type 2 Diabetes Mellitus |
Astellas Pharma Europe B.V.|Astellas Pharma Inc |
October 27 2011 | Phase 1 |
| NCT01611415 | Completed | Healthy Subjects|Pharmacokinetics of Ipragliflozin |
Astellas Pharma Europe B.V.|Astellas Pharma Inc |
July 2011 | Phase 1 |
| NCT01611428 | Completed | Bioavailability of Ipragliflozin|Healthy Subjects |
Astellas Pharma Europe B.V.|Astellas Pharma Inc |
June 2011 | Phase 1 |
Technischer Support
Tel: +1-832-582-8158 Ext:3
Wenn Sie weitere Fragen haben, hinterlassen Sie bitte eine Nachricht.
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.