Chk Inhibitoren | Chk Inhibitors
| Kat.-Nr. | Produktname | Informationen | Publikationen | Validierung |
|---|---|---|---|---|
| S1532 | AZD7762 | AZD7762 ist ein potenter und selektiver Inhibitor von Chk1 mit einem IC50 von 5 nM in einem zellfreien Assay. Es ist gleichermaßen potent gegen Chk2 und weniger potent gegen CAM, Yes, Fyn, Lyn, Hck und Lck. Phase 1. |
|

|
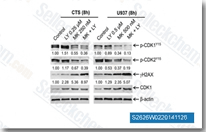
| S2626 | Rabusertib (LY2603618) | Rabusertib (LY2603618, IC-83) ist ein hochselektiver Chk1-Inhibitor mit potenzieller Antitumoraktivität in einem zellfreien Assay. IC50=7 nM, was eine etwa 100-fach höhere Wirksamkeit gegen Chk1 als gegen jede der anderen evaluierten Proteinkinasen zeigt. Rabusertib (LY2603618) induziert Zellzyklusarrest, DNA-Schadensantwort und autophagy in Krebszellen. Rabusertib (LY2603618) induziert Bak-abhängige apoptosis in AML-Zelllinien. |
|
|
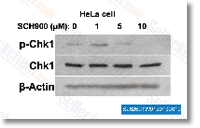
| S2735 | MK-8776 (SCH 900776) | MK-8776 (SCH 900776) ist ein selektiver Chk1-Inhibitor mit einem IC50 von 3 nM in einem zellfreien Assay, der eine 500-fache Selektivität gegenüber Chk2 aufweist. Er befindet sich in Phase 2. |
|
|
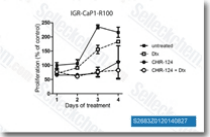
| S2683 | CHIR-124 | CHIR-124 ist ein neuartiger und potenter Chk1-Inhibitor mit einer IC50 von 0,3 nM in einem zellfreien Assay. Er zeigt eine 2.000-fache Selektivität gegenüber Chk2 und eine 500- bis 5.000-fach geringere Aktivität gegenüber CDK2/4 und Cdc2. |
|
|
| S7178 | Prexasertib (LY2606368) Dihydrochloride | Prexasertib HCl (LY2606368) ist ein ATP-kompetitiver CHK1-Inhibitor mit einem Ki-Wert von 0,9 nmol/L. Für CHK2 und RSK liegen die IC50-Werte in zellfreien Assays bei 8 nM bzw. 9 nM. |
|

|
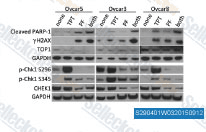
| S2904 | PF-477736 | PF-477736 (PF-736, PF-00477736) ist ein selektiver, potenter und ATP-kompetitiver Chk1-Inhibitor mit einem Ki von 0,49 nM in einem zellfreien Assay und hemmt auch VEGFR2, Aurora-A, FGFR3, Flt3, Fms (CSF1R), Ret und Yes. Es zeigt eine ~100-fache Selektivität für Chk1 gegenüber Chk2. |
|
|
| S6385 | Prexasertib (LY2606368) |
Prexasertib (LY2606368, ACR 368) ist ein selektiver ATP-kompetitiver Inhibitor von Chk1 und Chk2 mit IC50-Werten von 1 nM bzw. 8 nM in zellfreien Assays. Es hemmt auch RSK1 mit einer IC50 von 9 nM in zellfreien Assays. |
|
|
| S8632 | BML-277 (Chk2 Inhibitor II) | BML-277 (Chk2 Inhibitor II) ist ein ATP-kompetitiver Inhibitor von Chk2 mit einer IC50 von 15 nM und 1000-fach selektiver gegenüber Chk2-Serin/Threonin-Kinase als gegenüber Chk1- und Cdk1/B-Kinasen. Diese Verbindung schützt menschliche CD4(+)- und CD8(+)-T-Zellen dosisabhängig vor Apoptose durch ionisierende Strahlung. |
|
|
| S8253 | CCT245737 (SRA737) | CCT245737 (SRA737) ist ein oral aktiver CHK1-Inhibitor mit einer IC50 von 1,4 nM. Er zeigt eine >1.000-fache Selektivität gegenüber CHK2 und CDK1. |
|
|
| S8526 | GDC-0575 | GDC-0575 ist ein potenter und selektiver CHK1-Inhibitor mit einer IC50 von 1,2 nM. |
|
|
| E1991New | BBI-355 | BBI-355 ist ein oraler, potenter und selektiver niedermolekularer Inhibitor von CHK1 mit einem IC50-Wert von 0,3 nM. Er zeigt eine signifikante Antitumoraktivität als Einzelwirkstoff und in Kombination mit zielgerichteten Therapien in mehreren ecDNA+-Onkogen-amplifizierten Tumormodellen. | ||
| E1653 | CCT241533 hydrochloride | CCT241533 hydrochloride ist ein ATP-kompetitiver, potenter und selektiver Inhibitor von CHK2 mit einer IC50 von 3 nM und einem Ki von 1,16 nM und zeigt minimale Kreuzreaktivität gegen ein Panel von Kinasen. CCT241533 hemmt die CHK2-Aktivität in menschlichen Tumorzelllinien, wenn diese einer DNA-Schädigung ausgesetzt werden. | ||
| S7740 | SAR-020106 | SAR-020106 ist ein ATP-kompetitiver, potenter und selektiver CHK1-Inhibitor mit einer IC50 von 13,3 nM. | ||
| E5832New | BBI-2779 | BBI-2779 ist ein potenter, selektiver und oral aktiver Inhibitor von CHK1 mit einer IC50 von 0,3 nM. Er tötet bevorzugt Krebszellen ab, die extrachromosomale DNA (ecDNA) enthalten, indem er Replikationsstress und Zelltod induziert. In Magenkrebsmodellen mit FGFR2-Genamplifikation auf ecDNA unterdrückt BBI-2779 das Tumorwachstum und verhindert Arzneimittelresistenz. | ||
| E1667 | LY2880070 | LY2880070 ist ein oral aktiver Inhibitor von CHK1. Diese Verbindung kann als Antikrebsmittel eingesetzt werden und zeigt vielversprechende Ergebnisse in präklinischen Modellen des duktalen Pankreasadenokarzinoms (PDAC) in Kombination mit DNA-schädigenden Wirkstoffen. | ||
| S8148 | PD0166285 | PD0166285 ist ein potenter Wee1- und Chk1-Inhibitor mit Aktivität in nanomolaren Konzentrationen (IC50=24 nM für Wee1 und 72 nM für Myt1). Diese Verbindung ist auch ein neuartiger G2-Checkpoint-Abrogator. Sie induziert Apoptose. |
|
|
| S9639 | VX-803 (M4344) | VX-803 (M4344, ATR-Inhibitor 2) ist ein ATP-kompetitiver, oral aktiver und selektiver Inhibitor der Ataxia-telangiectasia- und Rad3-verwandten (ATR-)Kinase mit einem Ki von < 150 pM. Diese Verbindung hemmt stark die ATR-gesteuerte phosphorylierte Checkpoint-Kinase-1 (P-Chk1)-Phosphorylierung mit einer IC50 von 8 nM. Sie zeigt eine potenzielle antineoplastische Aktivität. |
|
|
| S6740 | DB07268 | DB07268 ist ein potenter und selektiver JNK1-Inhibitor mit einem IC50-Wert von 9 nM und zeigt eine mindestens 70- bis 90-fach höhere Potenz gegen JNK1 als CHK1, CK2 und PLK. |
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.