NF-κB controls the transcription of DNA. NF-κB is found in almost all animal cell types and is involved in cellular responses to stimuli such as stress, cytokines, free radicals, ultraviolet irradiation, oxidized LDL, and bacterial or viral antigens. [show the full text]
NF-κB Inhibitoren (NF-κB Inhibitors)
| Kat.-Nr. | Produktname | Informationen | Publikationen | Validierung |
|---|---|---|---|---|
| E4686 | DCZ0415 | DCZ0415 ist ein potenter Inhibitor von TRIP13. DCZ0415 beeinträchtigt die Reparatur durch nicht-homologes Endjoining und hemmt die NF-κB-Aktivität. Es löst Anti-Myelom-Effekte sowohl in vitro als auch in vivo und in primären Zellen aus, die von Myelompatienten gewonnen wurden, die gegen Medikamente resistent sind. |
|
|
| S7672 | Omaveloxolone (RTA-408) | Omaveloxolone (RTA-408) ist ein synthetisches Triterpenoid, das den zytoprotektiven Transkriptionsfaktor Nrf2 aktiviert und die NF-κB-Signalübertragung hemmt. Phase 2. |
|
|
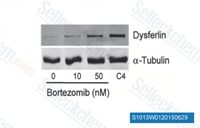
| S1013 | Bortezomib | Bortezomib ist ein potenter 20S Proteasome-Inhibitor mit einem Ki von 0,6 nM. Er zeigt eine günstige Selektivität gegenüber Tumorzellen im Vergleich zu normalen Zellen. Diese Verbindung hemmt NF-κB und induziert ERK-Phosphorylierung, um Cathepsin B zu unterdrücken und den katalytischen Prozess der Autophagy in Eierstockkrebs und anderen soliden Tumoren zu hemmen. |
|
|
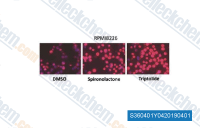
| S3604 | Triptolide | Triptolide ist ein Diterpen-Triepoxid, ein immunsuppressives Mittel, das aus der chinesischen Heilpflanze Tripterygium wilfordii extrahiert wird. Es fungiert als NF-κB-Inhibitor mit doppelter Wirkung durch Störung der p65/CBP-Interaktion und durch Reduktion des p65-Proteins. Triptolide (PG490) hebt die Transaktivierungsfunktion des Hitzeschock-Transkriptionsfaktors 1 (HSF1) auf. Triptolide hemmt MDM2 und induziert Apoptose über einen p53-unabhängigen Signalweg. |
|
|
| S8341 | TAK-243 (MLN7243) | TAK-243 (MLN7243) ist ein potenter, mechanismusbasierter niedermolekularer Inhibitor des ubiquitin activating enzyme (UAE) mit einer IC50 von 1 ± 0,2 nM im UBCH10 E2 Thioester-Assay. Es weist eine minimale Hemmaktivität in einer Reihe von Kinase- und Rezeptor-Assays sowie auf die menschliche Carboanhydrase Typ I und Typ II auf. TAK-243 (MLN7243) induziert ER stress, hebt die NF-κB-Signalwegaktivierung auf und fördert die apoptosis. |
|
|
| S8483 | CBL0137 Hydrochloride | CBL0137 (CBLC137, Curaxin 137) HCl aktiviert p53 und hemmt NF-κB mit EC50-Werten von 0,37 μM bzw. 0,47 μM in zellbasierten p53- und NF-kB-Reporterassays. Es hemmt auch das Histon-Chaperon FACT (facilitates chromatin transcription complex). |
|
|
| S8078 | Bardoxolone Methyl (RTA 402) | Bardoxolone Methyl (RTA 402, TP-155, NSC 713200, CDDO Methyl Ester, CDDO-Me) ist ein IKK-Inhibitor, der potente proapoptotische und entzündungshemmende Aktivitäten zeigt; Auch ein potenter Nrf2-Aktivator und Nuklearer Faktor-κB (NF-κB)-Inhibitor. Bardoxolone Methyl hebt die Ferroptosis auf. Bardoxolone Methyl induziert Apoptose und Autophagie in Krebszellen. |
|

|
| S1623 | N-Acetylcysteine (NAC chemical, N-Acetyl-L-Cysteine) | Acetylcystein (N-acetyl-l-cystein, NAC, N-Acetylcystein) ist ein ROS(reaktive Sauerstoffspezies)-Inhibitor, der die Aktivität von Proteasom-Inhibitoren antagonisiert. Es ist auch ein Inhibitor der Tumornekrosefaktor-Produktion. Acetylcystein (N-acetyl-l-cystein) unterdrückt die TNF-induzierte NF-κB-Aktivierung durch Hemmung von IκB-Kinasen. Acetylcystein (N-acetyl-l-cystein) induziert Apoptose über den mitochondrial abhängigen Signalweg. Acetylcystein (N-acetyl-l-cystein) hemmt Ferroptosis und Virusreplikation.Lösungen sind instabil und sollten frisch zubereitet werden. |
|

|
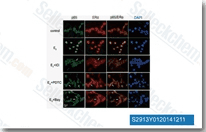
| S2913 | BAY 11-7082 (BAY 11-7821) | BAY 11-7082 (BAY 11-7821) ist ein NF-κB-Inhibitor, der die TNFα-induzierte IκBα-Phosphorylierung mit einem IC50 von 10 μM in Tumorzellen hemmt. BAY 11-7082 hemmt die Ubiquitin-spezifische Protease USP7 und USP21 mit einem IC50 von 0,19 μM bzw. 0,96 μM. BAY 11-7082 induziert Apoptose und S-Phasen-Arrest in Magenkrebszellen. |
|
|
| S7351 | JSH-23 | JSH-23 ist ein Inhibitor der NF-κB-Transkriptionsaktivität, der die LPS-stimulierte nukleäre Faktor (NF)-κB-Transkriptionsaktivität in RAW 264.7-Zellen mit einem IC50-Wert von 7,1 μM hemmt und die LPS-induzierte NF-κB-nukleäre Translokation ohne Beeinflussung des IκB-Abbaus stört. |
|

|
NF-κB (nuclear factor-kappa B) is a highly regulated, homo- or hetero-dimeric transcription factor, present in almost all cell types. The NF-κB proteins are composed of five different subunits, RelA (p65), RelB, c-Rel (Rel), NF-κB1, and NF-κB2, all of which share a Rel homology domain (RHD) in their N-termini, and have a transactivation domain in their C-termini, except for NF-κB1 and NF-κB2. The NF-κB1 and NF-κB2 proteins are synthesized as longer precursors, p105, and p100, which undergo selective degradation of their C-terminal region containing ankyrin repeats to generate the active NF-κB subunits, p50 and p52, respectively. [i] Different dimer combinations act as transcriptional activators or repressors, respectively. The p50 and p52 NF-κB members play critical roles in modulating the specificity of NF-κB function by forming heterodimers with RelA, RelB, or c-Rel. The NF-κB RelA-p50 and RelB-p50 heterodimeric complexes are transcriptional activators. The NF-κB p50/p50 and p52/p52 homodimers are generally transcriptional repressors, but can function as transcriptional activators when bound to nuclear protein Bcl-3. [2]
NF-κB is a rapidly acting primary transcription factor, and is controlled by subcellular compartmentalization and post-translational modifications (PTMs) including phosphorylation, acetylation, methylation and ubiquitylation. NF-κB dimers are primarily sequestered as an inactive form in the cytoplasm by a protein complex called inhibitor of kappa B (IκB) among unstimulated cells. Activation of NF-κB occurs via the degradation of IκB, a process initiated by IκB kinase (IKK). A variety of stimuli such as cytokines and cellular stress can activate the IKK, resulting in ubiquitination and dissociation of the IκB from NF-κB. The activated NF-κB is then translocated into the nucleus to regulate gene expression. NF-κB regulates a broad range of genes involved in various biological processes including inflammation, immunity, differentiation, development, as well as genes regulating cell proliferation, apoptosis, cell adhesion and the cellular microenviroment. In addition, NF-κB activates its own repressor IκBα and IκBε, as well as the TNFAIP3 (A20) a negative regulator of IKK activation, forming a negative feedback loop. [1]
NF-κB has been found to be constitutively active in a number of diseases, including arthritis, chronic inflammation, asthma, neurodegenerative diseases, and heart disease, as well as in many types of human tumors. [ii] NF-κB has long been linked with cancer, primarily through aberrant constitutive NF-κB activation that suppresses apoptosis or promotes tumor growth, metastasis, and angiogenesis by inducing the expression of anti-apoptotic genes, proto-oncogenes, matrix metalloproteinase, cell adhesion genes, and genes associated with the growth of new blood vessels. Additionally, NF-κB promotes a metabolic switch in cancer cells from oxidative phosphorylation to glycolysis (the Warburg effect) by inducing the expression of glycolytic enzymes and inhibiting the expression of mitochondrial gene. Constitutive activation of NF-κB can result from continuous exposure to NF-κB activating stimuli, such as cytokine release by tumor-associated macrophages (TAMs), or from mutations in NF-κB subunits and genes involved in regulating NF-κB function. Inhibiting NF-κB activation can prevent tumor cell proliferation and induce cell death. Given the importance of NF-κB in initiating or enhancing cell survival, NF-κB is therefore considered as a promising target for anticancer therapies. [1]
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.