nur für Forschungszwecke
Triptolide Hsp90 Middle Domain Inhibitor
Kat.-Nr.S3604
Chemische Struktur
Molekulargewicht: 360.4
Qualitätskontrolle
| Verwandte Ziele | NF-κB HDAC Antioxidant ROS IκB/IKK Nrf2 AP-1 MALT NOD |
|---|---|
| Weitere ADC Cytotoxin Inhibitoren | SN-38 Luteolin (+)-Bicuculline Rutin Artemisinin BHQ Pinocembrin Harmine hydrochloride Luteoloside Sacituzumab-govitecan |
Zellkultur, Behandlung & Arbeitskonzentration
| Zelllinien | Assay-Typ | Konzentration | Inkubationszeit | Formulierung | Aktivitätsbeschreibung | PMID |
|---|---|---|---|---|---|---|
| HT-29 | Cytotoxicity against | Cytotoxicity against human HT-29 cells, IC50=0.0021μM | 21470864 | |||
| HCT116 | Cytotoxicity against | Cytotoxicity against human HCT116 cells assessed as decrease in cell viability, IC50=0.0047μM | 31121546 | |||
| SKOV3 | Antiproliferative activity against | 72 hrs | Antiproliferative activity against human SKOV3 cells after 72 hrs by SRB assay, IC50=0.006μM | 20833543 | ||
| SKOV3 | Cytotoxicity against | 72 hrs | Cytotoxicity against human SKOV3 cells after 72 hrs by sulforhodamine B assay, IC50=0.006μM | 24378709 | ||
| SKOV3 | Cytotoxic activity against | 72 hrs | Cytotoxic activity against human SKOV3 cells assessed as reduction in cell viability after 72 hrs by SRB assay, IC50=0.0072μM | 28011223 | ||
| KBM5 | Cytotoxicity against | 72 hrs | Cytotoxicity against imatinib-resistant human KBM5 cells harboring Bcr-Abl T315I mutant after 72 hrs by MTS assay, IC50=0.0083μM | 20149665 | ||
| SKOV3 | Cytotoxicity against | Cytotoxicity against human SKOV3 cells by SRB assay, IC50=0.009μM | 19637874 | |||
| MDA-MB-468 | Cytotoxicity against | Cytotoxicity against human MDA-MB-468 cells by SRB assay, IC50=0.01μM | 19637874 | |||
| HCT116 | Cytotoxicity against | 72 hrs | Cytotoxicity against human HCT116 cells after 72 hrs by MTT assay, IC50=0.01μM | 19637874 | ||
| SKOV3 | Cytotoxicity against | 72 hrs | Cytotoxicity against human SKOV3 cells after 72 hrs by MTT assay, IC50=0.01μM | 19637874 | ||
| KBM5 | Cytotoxicity against | 72 hrs | Cytotoxicity against human KBM5 cells harboring wild type Bcr-Abl after 72 hrs by MTS assay, IC50=0.0103μM | 20149665 | ||
| Rh30 | Cytotoxicity against | 72 hrs | Cytotoxicity against human Rh30 cells after 72 hrs by MTT assay, IC50=0.014μM | 19637874 | ||
| A549 | Antagonist activity at | Antagonist activity at human PAR2 expressed in human A549 cells assessed as inhibition of 2f-LIGRLO-NH2-induced NFkappaB activation by luciferase reporter gene assay, IC50=0.014μM | 23895492 | |||
| SGC7901 | Cytotoxicity against | 72 hrs | Cytotoxicity against human SGC7901 cells after 72 hrs by MTT assay, IC50=0.015μM | 19637874 | ||
| MOLT4 | Cytotoxicity against | 72 hrs | Cytotoxicity against human MOLT4 cells after 72 hrs by MTT assay, IC50=0.017μM | 19637874 | ||
| A549 | Cytotoxic activity against | 72 hrs | Cytotoxic activity against human A549 cells assessed as reduction in cell viability after 72 hrs by SRB assay, IC50=0.0175μM | 28011223 | ||
| SMMC7721 | Cytotoxicity against | 72 hrs | Cytotoxicity against human SMMC7721 cells after 72 hrs by MTT assay, IC50=0.018μM | 19637874 | ||
| PC3 | Cytotoxic activity against | 72 hrs | Cytotoxic activity against human PC3 cells assessed as reduction in cell viability after 72 hrs by SRB assay, IC50=0.0183μM | 28011223 | ||
| MCF7 | Cytotoxicity against | 72 hrs | Cytotoxicity against human MCF7 cells after 72 hrs by MTT assay, IC50=0.019μM | 19637874 | ||
| A549 | Cytotoxicity against | Cytotoxicity against human A549 cells, IC50=0.019μM | 21470864 | |||
| PC3 | Cytotoxicity against | Cytotoxicity against human PC3 cells by SRB assay, IC50=0.02μM | 19637874 | |||
| Bel7402 | Cytotoxicity against | 72 hrs | Cytotoxicity against human Bel7402 cells after 72 hrs by MTT assay, IC50=0.02μM | 19637874 | ||
| PC3 | Antiproliferative activity against | 72 hrs | Antiproliferative activity against human PC3 cells after 72 hrs by SRB assay, IC50=0.02μM | 20833543 | ||
| PC3 | Cytotoxicity against | 72 hrs | Cytotoxicity against human PC3 cells after 72 hrs by sulforhodamine B assay, IC50=0.02μM | 24378709 | ||
| PC3 | Cytotoxicity against | Cytotoxicity against human PC3 cells assessed as inhibition of cell proliferation by sulforhodamine B assay, IC50=0.02μM | 25467158 | |||
| 786-O | Cytotoxicity against | 72 hrs | Cytotoxicity against human 786-O cells after 72 hrs by MTT assay, IC50=0.022μM | 19637874 | ||
| A549 | Antagonist activity at | Antagonist activity at human PAR2 expressed in human A549 cells coexpressing TACR1 assessed as inhibition of substance P-induced IL-8 production by ELISA, IC50=0.023μM | 23895492 | |||
| MDA-MB-231 | Cytotoxicity against | 72 hrs | Cytotoxicity against human MDA-MB-231 cells after 72 hrs by MTT assay, IC50=0.024μM | 19637874 | ||
| DU145 | Cytotoxicity against | 72 hrs | Cytotoxicity against human DU145 cells after 72 hrs by MTT assay, IC50=0.024μM | 19637874 | ||
| HO8910 | Cytotoxicity against | 72 hrs | Cytotoxicity against human HO8910 cells after 72 hrs by MTT assay, IC50=0.028μM | 19637874 | ||
| HCT15 | Cytotoxicity against | 72 hrs | Cytotoxicity against human HCT15 cells after 72 hrs by MTT assay, IC50=0.029μM | 19637874 | ||
| A549 | Growth inhibition of human | Growth inhibition of human A549 cells, IC50=0.03μM | 28814374 | |||
| 32D | Cytotoxicity against | 72 hrs | Cytotoxicity against mouse 32D cells harboring wild type Bcr-Abl after 72 hrs by MTS assay, IC50=0.032μM | 20149665 | ||
| U251 | Cytotoxicity against | Cytotoxicity against human U251 cells assessed as inhibition of cell proliferation by sulforhodamine B assay, IC50=0.033μM | 25467158 | |||
| 32D | Cytotoxicity against | 72 hrs | Cytotoxicity against imatinib-resistant mouse 32D cells harboring Bcr-Abl T315I mutant after 72 hrs by MTS assay, IC50=0.034μM | 20149665 | ||
| PC3 | Cytotoxicity against | 72 hrs | Cytotoxicity against human PC3 cells after 72 hrs by MTT assay, IC50=0.043μM | 19637874 | ||
| KB | Cytotoxicity against | 72 hrs | Cytotoxicity against human KB cells after 72 hrs by MTT assay, IC50=0.043μM | 19637874 | ||
| HepG2 | Cytotoxicity against | 48 hrs | Cytotoxicity against human HepG2 cells after 48 hrs by XTT assay, IC50=0.0433μM | 30613335 | ||
| HeLa | Cytotoxicity against | 72 hrs | Cytotoxicity against human HeLa cells after 72 hrs by MTT assay, IC50=0.047μM | 19637874 | ||
| U251 | Cytotoxicity against | 72 hrs | Cytotoxicity against human U251 cells after 72 hrs by MTT assay, IC50=0.049μM | 19637874 | ||
| K562 | Cytotoxicity against | 72 hrs | Cytotoxicity against human K562 cells after 72 hrs by MTT assay, IC50=0.05μM | 19637874 | ||
| NIH/3T3 | Cytotoxicity against | Cytotoxicity against mouse NIH/3T3 cells assessed as decrease in cell viability, IC50=0.05μM | 31121546 | |||
| SW1116 | Cytotoxicity against | 72 hrs | Cytotoxicity against human SW1116 cells after 72 hrs by MTT assay, IC50=0.052μM | 19637874 | ||
| A549 | Cytotoxicity against | 72 hrs | Cytotoxicity against human A549 cells after 72 hrs by MTT assay, IC50=0.059μM | 19637874 | ||
| HeLa | Cytotoxicity against | Cytotoxicity against human HeLa cells assessed as decrease in cell viability, IC50=0.087μM | 31121546 | |||
| Jurkat | Cytotoxicity against | Cytotoxicity against human Jurkat cells assessed as decrease in cell viability, IC50=0.14μM | 31121546 | |||
| MKN28 | Cytotoxicity against | 72 hrs | Cytotoxicity against human MKN28 cells after 72 hrs by MTT assay, IC50=0.2μM | 19637874 | ||
| MDCK | Cytotoxicity against | Cytotoxicity against MDCK cells assessed as decrease in cell viability, IC50=1.2μM | 31121546 | |||
| Pkd1-/- | Induction of | Induction of cell growth arrest in mouse Pkd1-/- cells in presence of calcium | 17360534 | |||
| Pkd1-/- | Increase in | 100 nM | 96 hrs | Increase in p21CIP/WAF expression in mouse Pkd1-/- cells at 100 nM after 96 hrs by Western blot analysis | 17360534 | |
| Pkd1+/- | Increase in | 100 nM | Increase in PC2 dependent calcium release in mouse Pkd1+/- cells at 100 nM | 17360534 | ||
| Pkd1-/- | Increase in | 100 nM | Increase in PC2 dependent calcium release in mouse Pkd1-/- cells at 100 nM | 17360534 | ||
| Pkd1-/- | Increase in | 50 uM | Increase in calcium release in mouse Pkd1-/- cells at 50 uM in presence of RyR antagonist dantrolene | 17360534 | ||
| Pkd2+/- | Growth inhibition of PC2 expressing mouse | 100 nM | 24 hrs | Growth inhibition of PC2 expressing mouse Pkd2+/- cells as cell death at 100 nM after 24 hrs | 17360534 | |
| KBM5 | Cytotoxicity against | 0.001 to 17 uM | 72 hrs | Cytotoxicity against human KBM5 cells harboring wild type Bcr-Abl at 0.001 to 17 uM after 72 hrs by MTS assay | 20149665 | |
| KBM5 | Cytotoxicity against | 0.001 to 17 uM | 72 hrs | Cytotoxicity against imatinib-resistant human KBM5 cells harboring Bcr-Abl T315I mutant at 0.001 to 17 uM after 72 hrs by MTS assay | 20149665 | |
| LNCAP | Antagonist activity at | 5 uM | 24 hrs | Antagonist activity at AR in human LNCAP cells assessed as suppression of DHT-induced receptor transcriptional activity at 5 uM after 24 hrs by dual luciferase reporter gene assay | 27994731 | |
| LNCAP | Antagonist activity at | 500 nM | 24 hrs | Antagonist activity at AR in human LNCAP cells assessed as suppression of DHT-induced receptor transcriptional activity at 500 nM after 24 hrs by dual luciferase reporter gene assay | 27994731 | |
| HepG2 | Antitumor activity against | 0.2 mg/kg | 15 days | Antitumor activity against human HepG2 cells xenografted in Balb/c nude mouse assessed as reduction in tumor growth at 0.2 mg/kg, ip administered once daily for 15 days | 30613335 | |
| Klicken Sie hier, um weitere experimentelle Daten zu Zelllinien anzuzeigen | ||||||
Chemische Informationen, Lagerung & Stabilität
| Molekulargewicht | 360.4 | Formel | C20H24O6 |
Lagerung (Ab dem Eingangsdatum) | |
|---|---|---|---|---|---|
| CAS-Nr. | 38748-32-2 | SDF herunterladen | Lagerung von Stammlösungen |
|
|
Löslichkeit
|
In vitro |
DMSO
: 72 mg/mL
(199.77 mM)
Water : Insoluble Ethanol : Insoluble |
Molaritätsrechner
|
In vivo |
|||||
In-vivo-Formulierungsrechner (Klare Lösung)
Schritt 1: Geben Sie die untenstehenden Informationen ein (Empfohlen: Ein zusätzliches Tier zur Berücksichtigung von Verlusten während des Experiments)
Schritt 2: Geben Sie die In-vivo-Formulierung ein (Dies ist nur der Rechner, keine Formulierung. Bitte kontaktieren Sie uns zuerst, wenn es im Abschnitt "Löslichkeit" keine In-vivo-Formulierung gibt.)
Berechnungsergebnisse:
Arbeitskonzentration: mg/ml;
Methode zur Herstellung der DMSO-Stammlösung: mg Wirkstoff vorgelöst in μL DMSO ( Konzentration der Stammlösung mg/mL, Bitte kontaktieren Sie uns zuerst, wenn die Konzentration die DMSO-Löslichkeit der Wirkstoffcharge überschreitet. )
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügenμL PEG300, mischen und klären, dann hinzufügenμL Tween 80, mischen und klären, dann hinzufügen μL ddH2O, mischen und klären.
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügen μL Maisöl, mischen und klären.
Hinweis: 1. Bitte stellen Sie sicher, dass die Flüssigkeit klar ist, bevor Sie das nächste Lösungsmittel hinzufügen.
2. Achten Sie darauf, das/die Lösungsmittel der Reihe nach hinzuzufügen. Sie müssen sicherstellen, dass die bei der vorherigen Zugabe erhaltene Lösung eine klare Lösung ist, bevor Sie mit der Zugabe des nächsten Lösungsmittels fortfahren. Physikalische Methoden wie Vortex, Ultraschall oder ein heißes Wasserbad können zur Unterstützung des Lösens verwendet werden.
Wirkmechanismus
| Targets/IC50/Ki |
NF-κB
HSF1
MDM2
|
|---|---|
| In vitro |
Triptolide ist ein Diterpen-Triepoxid mit potenten immunsuppressiven und entzündungshemmenden Eigenschaften. Diese Verbindung hemmt die Expression von IL-2 in aktivierten T-Zellen auf der Ebene der Purin-Box/Kernfaktor- und NF-κB-vermittelten Transkriptionsaktivierung. Es hemmt die Proliferation und Koloniebildung von Tumorzellen in extrem niedrigen Konzentrationen (2–10 ng/mL). Diese Chemikalie hat eine hemmende Wirkung auf Brust-, Magen- und Leukämie-Zelllinie HL-60-Zellen. Es induziert Apoptose in Tumorzellen durch Blockierung der NF-κB-Aktivierung und Sensibilisierung von Tumorzellen für TNF-&alpha-induzierten programmierten Zelltod.
|
| In vivo |
Triptolide wirkt synergistisch mit Cyclosporin A bei der Förderung des Transplantatüberlebens in Tiermodellen und bei der Unterdrückung der Graft-versus-Host-Krankheit bei allogenen Knochenmarktransplantationen. Darüber hinaus induziert es Apoptose in Tumorzellen und verstärkt die Tumornekrosefaktor (TNF-α)-Induktion der Apoptose, teilweise durch die Unterdrückung der c-IAP2- und c-IAP1-Induktion. Die Behandlung mit dieser Verbindung über 2–3 Wochen hemmt das Wachstum von Xenografts, die von vier verschiedenen Tumorzelllinien gebildet werden (B16-Melanom, MDA-435-Brustkrebs, TSU-Blasenkrebs und MGC80-3-Magenkarzinom), was darauf hindeutet, dass TPL ein breites Spektrum an Aktivitäten gegen Tumore aufweist, die sowohl Wildtyp- als auch mutierte Formen von p53 enthalten. Darüber hinaus hemmt es die experimentelle Metastasierung von B16F10-Zellen in die Lungen und Milzen von Mäusen. Es hat In-vitro- und In-vivo-Aktivitäten gegen Mausmodelle der polyzystischen Nierenerkrankung. LD50: Mäuse 0,83 mg/kg (i.v.).
|
Literatur |
|
Anwendungen
| Methoden | Biomarker | Bilder | PMID |
|---|---|---|---|
| Western blot | c-Jun MDM2 p-AKT / AKT / p-Foxo3a / Foxo3a / p53 p-PI3K / PI3K / p85 / p110 |
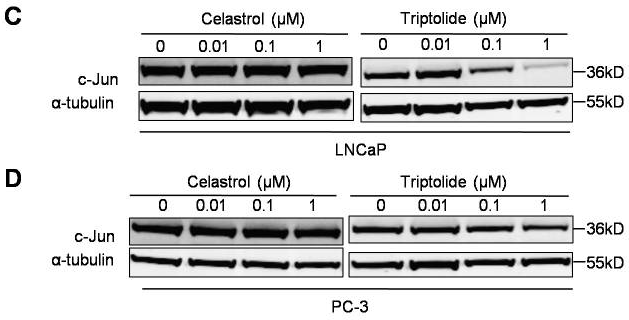
|
22666381 |
| Growth inhibition assay | Cell viability |
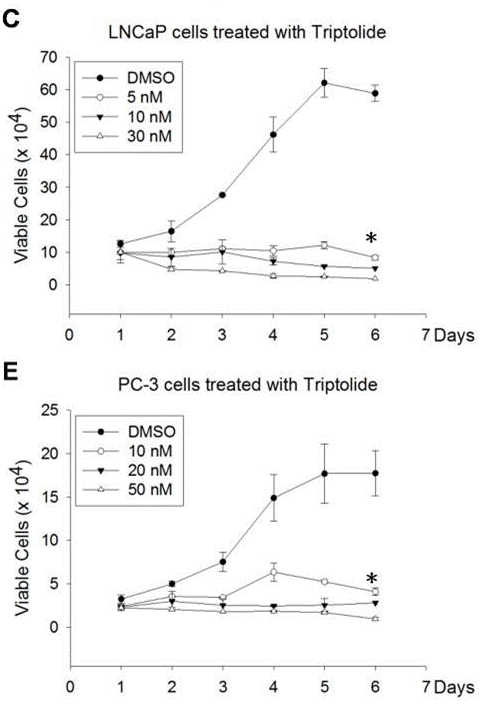
|
22666381 |
Technischer Support
Tel: +1-832-582-8158 Ext:3
Wenn Sie weitere Fragen haben, hinterlassen Sie bitte eine Nachricht.
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.