nur für Forschungszwecke
Safinamide Mesylate MAO Inhibitor
Kat.-Nr.S1472
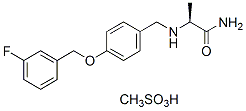
Chemische Struktur
Molekulargewicht: 398.45
Qualitätskontrolle
- In Nature Medicine für seine erstklassige Qualität zitiert
- COA
- NMR
- HPLC
- SDS
- Datenblatt
| Verwandte Ziele | Dehydrogenase HSP Transferase P450 (e.g. CYP17) PDE phosphatase PPAR Vitamin Carbohydrate Metabolism Mitochondrial Metabolism |
|---|---|
| Weitere MAO Inhibitoren | Moclobemide (Ro 111163) Sennoside A Paeonol Isatin Hypericin J147 Amezinium (methylsulfate) Xanthoangelol Norharmane Myristicin |
Chemische Informationen, Lagerung & Stabilität
| Molekulargewicht | 398.45 | Formel | C17H19FN2O2.CH4O3S |
Lagerung (Ab dem Eingangsdatum) | |
|---|---|---|---|---|---|
| CAS-Nr. | 202825-46-5 | SDF herunterladen | Lagerung von Stammlösungen |
|
|
| Synonyme | PNU-151774E,FCE28073 | Smiles | CC(C(=O)N)NCC1=CC=C(C=C1)OCC2=CC(=CC=C2)F.CS(=O)(=O)O | ||
Löslichkeit
|
In vitro |
DMSO
: 80 mg/mL
(200.77 mM)
Water : 80 mg/mL Ethanol : 13 mg/mL |
Molaritätsrechner
|
In vivo |
|||||
In-vivo-Formulierungsrechner (Klare Lösung)
Schritt 1: Geben Sie die untenstehenden Informationen ein (Empfohlen: Ein zusätzliches Tier zur Berücksichtigung von Verlusten während des Experiments)
Schritt 2: Geben Sie die In-vivo-Formulierung ein (Dies ist nur der Rechner, keine Formulierung. Bitte kontaktieren Sie uns zuerst, wenn es im Abschnitt "Löslichkeit" keine In-vivo-Formulierung gibt.)
Berechnungsergebnisse:
Arbeitskonzentration: mg/ml;
Methode zur Herstellung der DMSO-Stammlösung: mg Wirkstoff vorgelöst in μL DMSO ( Konzentration der Stammlösung mg/mL, Bitte kontaktieren Sie uns zuerst, wenn die Konzentration die DMSO-Löslichkeit der Wirkstoffcharge überschreitet. )
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügenμL PEG300, mischen und klären, dann hinzufügenμL Tween 80, mischen und klären, dann hinzufügen μL ddH2O, mischen und klären.
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügen μL Maisöl, mischen und klären.
Hinweis: 1. Bitte stellen Sie sicher, dass die Flüssigkeit klar ist, bevor Sie das nächste Lösungsmittel hinzufügen.
2. Achten Sie darauf, das/die Lösungsmittel der Reihe nach hinzuzufügen. Sie müssen sicherstellen, dass die bei der vorherigen Zugabe erhaltene Lösung eine klare Lösung ist, bevor Sie mit der Zugabe des nächsten Lösungsmittels fortfahren. Physikalische Methoden wie Vortex, Ultraschall oder ein heißes Wasserbad können zur Unterstützung des Lösens verwendet werden.
Wirkmechanismus
| Merkmale |
Greater than 5,000-fold potency in inhibiting MAO-B vs. MAO-A.
|
|---|---|
| Targets/IC50/Ki |
MAO-B
98 nM
|
| In vitro |
Safinamide ist ein hochselektiver MAO-B-Inhibitor in Rattenhirnmitochondrien mit einer IC50 von 98 nM. Safinamide hemmt MAO-B im menschlichen Gehirn mit einer IC50 von 9 nM. Safinamide hat eine hohe Affinität zur Na+-Kanal-Bindungsstelle II in Rattenkortikalmembranen mit einer IC50 von 8 μM. Safinamide hemmt die schnellen Na+-Ströme konzentrations- und zustandsabhängig in Rattenkortikalneuronen. Safinamide blockiert N-Typ-Ca2+-Ströme in Rattenkortikalneuronen mit einer IC50 von 23 μM. Safinamide hemmt die durch depolarisierende Bedingungen induzierte Glutamatfreisetzung in Rattenhippocampus-Synaptosomen mit einer IC50 von 9 μM. Safinamide, 1 Stunde vor Veratridin inkubiert, reduziert die Neuronschädigung mit einer IC50 von 1,4 μM durch Blockade der Öffnung spannungsabhängiger Na+- und Ca2+-Kanäle in primären kortikalen Rattenneuronen. Safinamide bindet an menschliche MAO B mit einem Ki von 0,5 μM. Safinamide bindet an menschliche MAO B in einer erweiterten Konformation, die sowohl die Flavin- als auch die Eintrittskavität besetzt.
|
| In vivo |
Safinamide, oral verabreicht, hemmt dosisabhängig die MAO-B im Gehirn von Mäusen mit einer IC50 von 0,6 mg/kg, und die MAO-B-Aktivität erholt sich schnell, beginnend nach 8 Stunden. Safinamide hemmt signifikant die Degeneration des Zellkörpers in der Substantia nigra pars compacta. Safinamide, 15 Minuten vor Kaininsäure intraperitoneal verabreicht, schützt vor dem Verlust von Hippocampusneuronen, beginnend bei 10 mg/kg, und zeigt neuroprotektive Eigenschaften. Safinamide, intraperitoneal in einer Dosis von 100 mg/kg verabreicht, zeigt eine relevante neurorettende Wirkung auf Hippocampusneuronen, wenn es 3 Stunden nach Ischämie gegeben wird. Safinamide hat eine hohe orale Bioverfügbarkeit (80-92%), wird schnell im Plasma resorbiert, erreicht den Höhepunkt innerhalb von 0,5-2 Stunden und fällt dann ab, mit einer terminalen Halbwertszeit von etwa 3, 7 bzw. 13 Stunden bei Mäusen, Ratten und Affen.
|
Literatur |
|
Technischer Support
Tel: +1-832-582-8158 Ext:3
Wenn Sie weitere Fragen haben, hinterlassen Sie bitte eine Nachricht.
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.