nur für Forschungszwecke
Fenebrutinib (GDC-0853) BTK Inhibitor
Kat.-Nr.S8421
Chemische Struktur
Molekulargewicht: 664.80
Qualitätskontrolle
- In Nature Medicine für seine erstklassige Qualität zitiert
- COA
- NMR
- SDS
- Datenblatt
| Verwandte Ziele | EGFR VEGFR PDGFR FGFR c-Met Src MEK CSF-1R FLT3 HER2 |
|---|---|
| Weitere BTK Inhibitoren | Catadegbrutinib (BGB-16673) Spebrutinib (AVL-292) tirabrutinib(ONO-4059) hydrochloride CGI1746 LFM-A13 CNX-774 Evobrutinib BMS-935177 Branebrutinib (BMS-986195) Pirtobrutinib (LOXO-305) |
Chemische Informationen, Lagerung & Stabilität
| Molekulargewicht | 664.80 | Formel | C37H44N8O4 |
Lagerung (Ab dem Eingangsdatum) | 3 years -20°C powder |
|---|---|---|---|---|---|
| CAS-Nr. | 1434048-34-6 | -- | Lagerung von Stammlösungen |
|
|
| Synonyme | N/A | Smiles | CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8 | ||
Löslichkeit
|
In vitro |
DMSO
: 8 mg/mL
(12.03 mM)
Water : Insoluble Ethanol : Insoluble |
Molaritätsrechner
|
In vivo |
|||||
In-vivo-Formulierungsrechner (Klare Lösung)
Schritt 1: Geben Sie die untenstehenden Informationen ein (Empfohlen: Ein zusätzliches Tier zur Berücksichtigung von Verlusten während des Experiments)
Schritt 2: Geben Sie die In-vivo-Formulierung ein (Dies ist nur der Rechner, keine Formulierung. Bitte kontaktieren Sie uns zuerst, wenn es im Abschnitt "Löslichkeit" keine In-vivo-Formulierung gibt.)
Berechnungsergebnisse:
Arbeitskonzentration: mg/ml;
Methode zur Herstellung der DMSO-Stammlösung: mg Wirkstoff vorgelöst in μL DMSO ( Konzentration der Stammlösung mg/mL, Bitte kontaktieren Sie uns zuerst, wenn die Konzentration die DMSO-Löslichkeit der Wirkstoffcharge überschreitet. )
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügenμL PEG300, mischen und klären, dann hinzufügenμL Tween 80, mischen und klären, dann hinzufügen μL ddH2O, mischen und klären.
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügen μL Maisöl, mischen und klären.
Hinweis: 1. Bitte stellen Sie sicher, dass die Flüssigkeit klar ist, bevor Sie das nächste Lösungsmittel hinzufügen.
2. Achten Sie darauf, das/die Lösungsmittel der Reihe nach hinzuzufügen. Sie müssen sicherstellen, dass die bei der vorherigen Zugabe erhaltene Lösung eine klare Lösung ist, bevor Sie mit der Zugabe des nächsten Lösungsmittels fortfahren. Physikalische Methoden wie Vortex, Ultraschall oder ein heißes Wasserbad können zur Unterstützung des Lösens verwendet werden.
Wirkmechanismus
| Targets/IC50/Ki |
BTK
(Cell-free assay) 0.91 nM(Ki)
BTK C481R
(Cell-free assay) 1.3 nM(Ki)
BTK C481S
(Cell-free assay) 1.6 nM(Ki)
BTK T474M
(Cell-free assay) 3.4 nM(Ki)
BTK T474I
(Cell-free assay) 12.6 nM(Ki)
|
|---|---|
| In vitro |
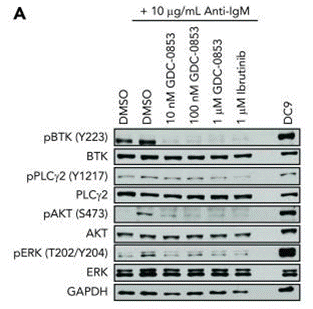
Bei Tests mit 1 μM gegen eine breite Palette humaner Kinase-biochemischer Assays hemmt Fenebrutinib (GDC-0853) nur 3 von 286 Off-Target-Kinasen. Basierend auf den bestimmten IC50-Werten beträgt die Selektivität für Btk >100-fach gegenüber jedem dieser 3 Off-Targets: Bmx (153-fach), Fgr (168-fach) und Src (131-fach). Diese Verbindung blockiert sowohl die B-Zell-BCR- als auch die Monozyten-FcγR-Signalisierung. Im In-vitro-biochemischen Btk-Enzymassay zeigt sie eine durchschnittliche Verweilzeit mit Btk von 18,3 ± 2,8 Stunden. Sie blockiert die zelluläre Autophosphorylierung von WT Btk und der C481S-Mutante. CLL-Zellen (chronisch lymphatische Leukämie), die in vitro vor der BCR-Stimulation mit GDC-0853 behandelt wurden, zeigen reduzierte BTK-Phosphorylierungslevel und eine verminderte Aktivierung von Downstream-Zielen, einschließlich PLCγ2, AKT und ERK. Es hemmt die NF-κB-abhängige Transkription, reduziert die Aktivierung und beeinträchtigt die Migration. Diese Verbindung hemmt EGFR und ITK im zellulären System nicht und beeinflusst die T-Zell-Rezeptor-Aktivierung nicht. |
| Kinase-Assay |
Kinase-Selektivität
|
|
Die Kinase-Selektivität von Fenebrutinib (GDC-0853) wird bei einer Konzentration von 1 µM in einem Panel von bis zu 287 rekombinanten humanen Kinase-Aktivitäts- und Bindungsassays bewertet, einschließlich zytoplasmatischer und Rezeptor-Tyrosinkinasen, Serin/Threonin-Kinasen und Lipidkinasen. Die Kinase-Aktivitätsassays messen die Peptidphosphorylierung oder ADP-Produktion, während die Bindungsassays die Verdrängung von ATP-Stelle-bindenden Sonden überwachen. Die in den Aktivitätsassays verwendeten ATP-Konzentrationen liegen typischerweise innerhalb des 2-fachen des experimentell bestimmten scheinbaren Michaeliskonstanten (Kmapp)-Werts für jede Kinase, während die in den Bindungsassays verwendeten kompetitiven Bindungs-Tracer-Konzentrationen im Allgemeinen innerhalb des 3-fachen der experimentell bestimmten Dissoziationskonstanten (Kd)-Werte liegen. Diese Verbindung wird dupliziert gegen jede Kinase getestet und die mittleren %-Inhibitionswerte werden berichtet. Für Kinasen, die bei der Testkonzentration um nahezu oder mehr als 80 % gehemmt werden, werden 10-Punkt-Inhibitor-Titrationen unter Verwendung derselben Assays durchgeführt, um die Inhibitor-Konzentrationen zu bestimmen, die eine 50 %ige Hemmung (IC50) verursachten.
|
|
| In vivo |
Fenebrutinib (GDC-0853) weist eine moderate Clearance von 27,4 mL/min/kg und eine ausgezeichnete Bioverfügbarkeit (F=65 %) bei Ratten auf, denen 0,2 mg/kg per intraperitonealer Injektion oder 1 mg/kg PO verabreicht wurden. Die Plasma-Clearance beträgt 27,4 mL/min/kg, das Verteilungsvolumen (Vd) 5,42 L/kg und die Plasmahalbwertszeit (t1/2) 2,2 h. Diese Verbindung zeigt auch günstige PK-Eigenschaften bei Hunden. Die Halbwertszeit von 3,8 Stunden (Clp 10,9 mL/min/kg, Vd 2,96 L/kg) und die hohe orale Bioverfügbarkeit (85 %) ermöglichen auch das Erreichen ausreichender Expositionen in toxikologischen Studien an Hunden. Es wird sowohl bei Ratten als auch bei Hunden gut vertragen und weist ein insgesamt günstiges Sicherheitsprofil auf. GDC-0853 ist nützlich bei der Behandlung von rheumatoider Arthritis und anderen B-Zell- oder myeloischen Zell-vermittelten Autoimmunerkrankungen. In einer Einzel-Dosis-Eskalationsstudie (SAD) (0,5 mg bis 600 mg) und einer Mehrfach-Dosis-Eskalationsstudie (MAD) über 14 Tage (250 mg BID bis 500 mg QD) wurde es sehr gut vertragen, ohne schwerwiegende unerwünschte Ereignisse, Sicherheitssignale oder dosislimitierende Toxizitäten. Es wurde gut resorbiert und hatte eine lineare, dosisproportionale Pharmakokinetik. Bei Sprague-Dawley (SD)-Ratten verursachte die Verabreichung dieser Verbindung und anderer strukturell diverser BTK-Inhibitoren über 7 Tage oder länger Pankreasläsionen, die aus multifokalen Insel-zentrierten Blutungen, Entzündungen, Fibrose und pigmentbeladenen Makrophagen mit angrenzender lobulärer exokriner Azinuszellatrophie, Degeneration und Entzündung bestanden. Ähnliche Befunde wurden bei Mäusen oder Hunden bei viel höheren Expositionen nicht beobachtet. |
Literatur |
|
Anwendungen
| Methoden | Biomarker | Bilder | PMID |
|---|---|---|---|
| Western blot | p-BTK / BTK / p-PLCγ2 / PLCγ2 / p-AKT / AKT / p-ERK / ERK |
|
30018078 |
Klinische Studieninformationen
(Daten von https://clinicaltrials.gov, aktualisiert am 2024-05-22)
| NCT-Nummer | Rekrutierung | Erkrankungen | Sponsor/Kooperationspartner | Startdatum | Phasen |
|---|---|---|---|---|---|
| NCT05119569 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 1 2022 | Phase 2 |
| NCT04586023 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 24 2021 | Phase 3 |
| NCT04586010 | Active not recruiting | Relapsing Multiple Sclerosis |
Hoffmann-La Roche |
March 17 2021 | Phase 3 |
| NCT03693625 | Terminated | Urticaria |
Genentech Inc. |
September 27 2018 | Phase 2 |
| NCT03596632 | Completed | Healthy Participants |
Hoffmann-La Roche |
July 27 2018 | Phase 1 |
Technischer Support
Tel: +1-832-582-8158 Ext:3
Wenn Sie weitere Fragen haben, hinterlassen Sie bitte eine Nachricht.
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.