nur für Forschungszwecke
Anlotinib (AL3818) Dihydrochloride VEGFR2 Inhibitor
Kat.-Nr.S8726
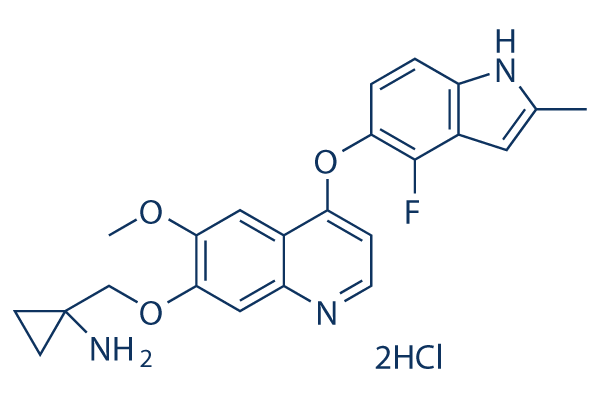
Chemische Struktur
Molekulargewicht: 480.36
Springe zu
Qualitätskontrolle
Charge:
Reinheit:
99.93%
99.93
| Verwandte Ziele | EGFR PDGFR FGFR c-Met Src MEK CSF-1R FLT3 HER2 c-Kit |
|---|---|
| Weitere VEGFR Inhibitoren | SAR131675 SU 5402 Cediranib (AZD2171) Vatalanib (PTK787) 2HCl Linifanib (ABT-869) Apatinib (YN968D1) Apatinib (YN968D1) mesylate Ki8751 ZM 323881 HCl Semaxanib (SU5416) |
Zellkultur, Behandlung & Arbeitskonzentration
| Zelllinien | Assay-Typ | Konzentration | Inkubationszeit | Formulierung | Aktivitätsbeschreibung | PMID |
|---|---|---|---|---|---|---|
| U-87MG | Function assay | 0.01, 0.1, 1, 10 and 100 μM | 1.5 h | inhibited PDGF-BB-stimulated phosphorylation of PDGFRβ, AKT and ERK in U-87MG cells | 29446853 | |
| Mo7e | Function assay | 0.01, 0.1, 1, 10 and 100 μM | 1.5 h | Anlotinib inhibited SCF‐1‐stimulated phosphorylation of c‐Kit, AKT and ERK in Mo7e cells | 29446853 | |
| HUVEC | Function assay | 0.01, 0.1, 1, 10 and 100 μM | 1.5 h | anlotinib inhibited VEGF‐stimulated intracellular phosphorylation of VEGFR2 in a concentration‐dependent way in HUVEC with a subnanomolar IC50 value | 29446853 | |
| A549 | Cell viability assay | 24, 48 and 72 h | IC50=64.82 μM(t=24 h); IC50=30.34 μM(t=48 h); IC50=17.29 μM(t=72 h) | 30755242 | ||
| NCI-H1975 | Cell viability assay | 8 μg/ml | 24 h | Cell viability was decreased remarkably | 30871526 | |
| Calu-1 | Cell viability assay | 24, 48 and 72 h | IC50=61.23 μM(t=24 h); IC50=36.8 μM(t=48 h); IC50=28.64 μM(t=72 h) | 30755242 | ||
| Klicken Sie hier, um weitere experimentelle Daten zu Zelllinien anzuzeigen | ||||||
Chemische Informationen, Lagerung & Stabilität
| Molekulargewicht | 480.36 | Formel | C23H22FN3O3.2HCl |
Lagerung (Ab dem Eingangsdatum) | 3 years -20°C powder |
|---|---|---|---|---|---|
| CAS-Nr. | 1360460-82-7 | -- | Lagerung von Stammlösungen |
|
|
| Synonyme | N/A | Smiles | CC1=CC2=C(N1)C=CC(=C2F)OC3=C4C=C(C(=CC4=NC=C3)OCC5(CC5)N)OC.Cl.Cl | ||
Löslichkeit
|
In vitro |
Water : 96 mg/mL
DMSO
: 48 mg/mL
(99.92 mM)
Ethanol : Insoluble |
Molaritätsrechner
Verdünnungsrechner
Molekulargewichtsrechner
|
In vivo |
|||||
In-vivo-Formulierungsrechner (Klare Lösung)
Schritt 1: Geben Sie die untenstehenden Informationen ein (Empfohlen: Ein zusätzliches Tier zur Berücksichtigung von Verlusten während des Experiments)
mg/kg
g
μL
Schritt 2: Geben Sie die In-vivo-Formulierung ein (Dies ist nur der Rechner, keine Formulierung. Bitte kontaktieren Sie uns zuerst, wenn es im Abschnitt "Löslichkeit" keine In-vivo-Formulierung gibt.)
% DMSO
%
% Tween 80
% ddH2O
%DMSO
%
Berechnungsergebnisse:
Arbeitskonzentration: mg/ml;
Methode zur Herstellung der DMSO-Stammlösung: mg Wirkstoff vorgelöst in μL DMSO ( Konzentration der Stammlösung mg/mL, Bitte kontaktieren Sie uns zuerst, wenn die Konzentration die DMSO-Löslichkeit der Wirkstoffcharge überschreitet. )
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügenμL PEG300, mischen und klären, dann hinzufügenμL Tween 80, mischen und klären, dann hinzufügen μL ddH2O, mischen und klären.
Methode zur Herstellung der In-vivo-Formulierung: Nehmen Sie μL DMSO Stammlösung, dann hinzufügen μL Maisöl, mischen und klären.
Hinweis: 1. Bitte stellen Sie sicher, dass die Flüssigkeit klar ist, bevor Sie das nächste Lösungsmittel hinzufügen.
2. Achten Sie darauf, das/die Lösungsmittel der Reihe nach hinzuzufügen. Sie müssen sicherstellen, dass die bei der vorherigen Zugabe erhaltene Lösung eine klare Lösung ist, bevor Sie mit der Zugabe des nächsten Lösungsmittels fortfahren. Physikalische Methoden wie Vortex, Ultraschall oder ein heißes Wasserbad können zur Unterstützung des Lösens verwendet werden.
Wirkmechanismus
| Targets/IC50/Ki |
VEGFR2
(Cell-free assay) 0.2 nM
VEGFR3
(Cell-free assay) 0.7 nM
c-Kit
(Cell-free assay) 14.8 nM
c-Kit
(Cell-free assay) 14.8 nM
c-Kit
(Cell-free assay) 14.8 nM
|
|---|---|
| In vitro |
Anlotinib besetzt die ATP-Bindungstasche der VEGFR2 Protein Tyrosine Kinase und zeigt eine hohe Selektivität und Hemmpotenz (IC50 <1 nmol/L) für VEGFR2 im Vergleich zu anderen Protein Tyrosine Kinases. Anlotinib hemmt VEGFR2 und VEGFR3 mit IC50-Werten von 0,2 bzw. 0,7 nmol/L. Die Hemmwirkung von Anlotinib gegen VEGFR1 ist geringer, mit einem IC50-Wert von 26,9 nmol/L. Die IC50-Werte von Anlotinib zur Hemmung der PDGFR-verwandten Kinasen c-Kit und PDGFRβ betragen 14,8 bzw. 115,0 nmol/L. Anlotinib hat selbst bei einer Konzentration von 2000 nmol/L wenig Einfluss auf die Aktivität anderer Kinasen, einschließlich c-Met, c-Src, EGFR und HER2. Anlotinib hemmt VEGF-induzierte Signalgebung und Zellproliferation in HUVEC mit picomolaren IC50-Werten. Mikromolare Konzentrationen von Anlotinib sind jedoch erforderlich, um die Tumorzellproliferation direkt in vitro zu hemmen. Anlotinib hemmt signifikant die HUVEC-Migration und Röhrchenbildung; es hemmt auch das Mikrogewäßwachstum aus Explantaten der Rattenaorta in vitro.
|
| Kinase-Assay |
Enzymimmunoassays
|
|
Die hemmende Wirkung von Anlotinib auf Protein Tyrosine Kinases wurde mittels ELISA bestimmt. Die Reaktion von ATP mit Protein Tyrosine Kinase wurde in Reaktionspuffer (50 mmol/L HEPES pH 7,4, 50 mmol/L MgCl2, 0,5 mmol/L MnCl2, 0,2 mmol/L Na3VO4, 1 mmol/L DTT) gestartet und 1 Stunde bei 37 °C in 96-Well-Platten inkubiert, die mit 20 μg/mL Poly(Glu,Tyr)4:1 vorbeschichtet waren. Die Platte wurde mit PY99-Antikörper und dann mit HRP-konjugiertem Anti-Maus-IgG inkubiert. Nach der Reaktion mit o-Phenylendiamin-Lösung und anschließender Beendigung durch Zugabe von 2N H2SO4 wurde die Absorption bei 490 nm mit einem Synergy H4 Hybrid-Reader gemessen.
|
|
| In vivo |
Anlotinib verringert die Gefäßdichte im Tumorgewebe in vivo. Im Vergleich zum bekannten Protein Tyrosine Kinase-Inhibitor Sunitinib zeigt eine einmal tägliche orale Dosis von Anlotinib eine breitere und stärkere in vivo-Antitumorwirksamkeit und führt in einigen Modellen zur Tumorregression bei Nacktmäusen. Es wird in Mäusen gut vertragen. Anlotinib ist in Dosen (1,5–6 mg/kg täglich) wirksam, die signifikant niedriger sind als die wirksamen Dosen anderer TKI, die Dosen von 20–100 mg/kg erfordern, um eine signifikante Hemmung des Tumorwachstums bei Mäusen zu erreichen. In vivo zeigte Anlotinib während der Dosierungsperiode eine breite Aktivität gegen humane Tumorzell-Xenograft-Modelle des Kolons (SW-620), der Eierstöcke (SK-OV-3), der Leber (SMMC-7721), der Niere (Caki-1), des Glioms (U87MG) und des nicht-kleinzelligen Lungenkarzinoms (Calu-3). Bei Sprague-Dawley-Ratten und Beagle-Hunden wird Anlotinib nach oraler Verabreichung schnell aus dem Magen-Darm-Trakt resorbiert. Die orale Bioverfügbarkeit beträgt 23-45 % bei Ratten und 47-74 % bei Hunden. Anlotinib weist bei beiden Spezies ein großes Verteilungsvolumen auf. Bei Ratten zeigen primäre Gewebe wie Lunge, Nieren, Leber und Herz signifikant höhere Anlotinib-Expositionsniveaus im Vergleich zu denen im Plasma. Das Expositionsniveau im Gehirn ist vergleichbar mit dem entsprechenden Plasmaspiegel. Bei tumortragenden Mäusen konzentriert sich Anlotinib im Tumorgewebe 2,4- bis 2,6-mal stärker als im Plasma. Beim Menschen weist Anlotinib eine recht lange t1/2 (96 ± 17 h) auf, die dosisunabhängig zu sein schien. Die terminale Halbwertszeit von Anlotinib bei Hunden (22,8±11,0 h) ist länger als die bei Ratten (5,1±1,6 h). Dieser Unterschied scheint hauptsächlich mit einem interspezifischen Unterschied in der totalen Plasmaclearance (Ratten, 5,35±1,31 L/h/kg; Hunde, 0,40±0,06 L/h/kg) verbunden zu sein. Im menschlichen Plasma ist Anlotinib überwiegend an Albumin und Lipoproteine gebunden, und nicht an α1-saures Glykoprotein oder γ-Globuline.
|
Literatur |
|
Anwendungen
| Methoden | Biomarker | Bilder | PMID |
|---|---|---|---|
| Western blot | TP53 / Cleaved-caspase 3 / Cleaved PARP Beclin-1 / LC3-I / LC3-II Akt / p-Akt / mTOR / p-mTOR |
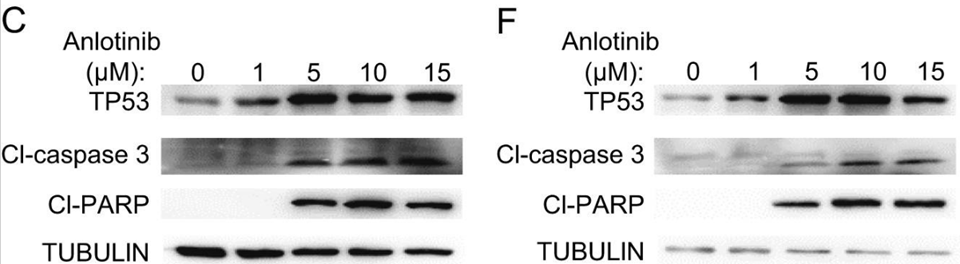
|
30139768 |
| Immunofluorescence | LC3-II |
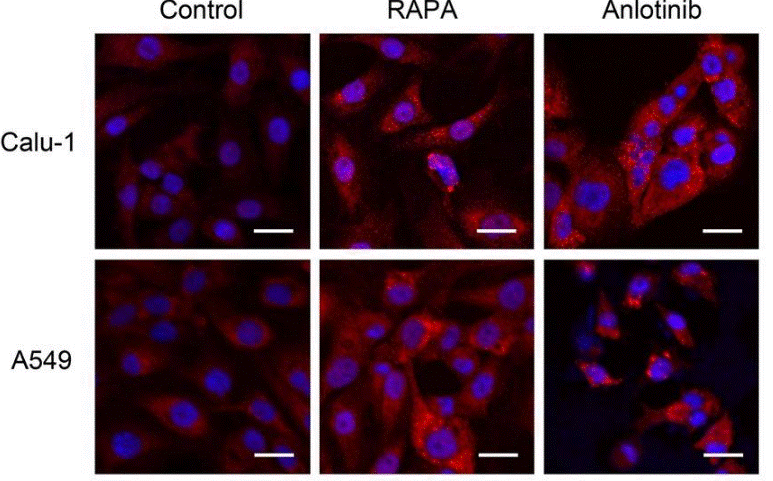
|
30755242 |
| Growth inhibition assay | Cell viability |
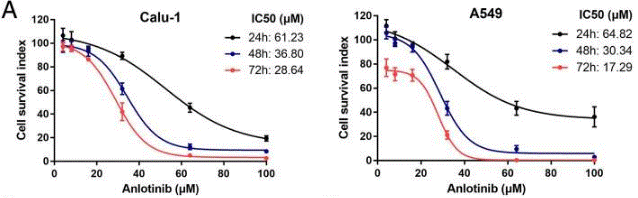
|
30755242 |
Klinische Studieninformationen
(Daten von https://clinicaltrials.gov, aktualisiert am 2024-05-22)
| NCT-Nummer | Rekrutierung | Erkrankungen | Sponsor/Kooperationspartner | Startdatum | Phasen |
|---|---|---|---|---|---|
| NCT06331169 | Not yet recruiting | Breast Cancer |
Fudan University |
April 1 2024 | Phase 1 |
| NCT06015061 | Recruiting | Pheochromocytoma Metastatic|Ultrasonography|Paraganglioma Malignant|Pheochromocytoma Malignant |
Peking Union Medical College Hospital |
March 1 2023 | -- |
| NCT05816499 | Recruiting | NSCLC Stage IV|NSCLC Stage IIIB|NSCLC Stage IIIC |
Shanghai Chest Hospital|The Affiliated Hospital of Qingdao University|Anhui Provincial Hospital|Zhejiang University |
February 16 2023 | Phase 1|Phase 2 |
| NCT05883085 | Recruiting | Pheochromocytoma|Paraganglioma |
Peking Union Medical College Hospital |
May 1 2022 | Phase 2 |
| NCT05301764 | Recruiting | Soft Tissue Sarcoma |
Lyvgen Biopharma Holdings Limited |
May 25 2022 | Phase 1|Phase 2 |
Technischer Support
Tel: +1-832-582-8158 Ext:3
Wenn Sie weitere Fragen haben, hinterlassen Sie bitte eine Nachricht.
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.