AKT inhibitors represent a pivotal class of targeted therapeutic agents that have revolutionized the landscape of cancer research and treatment. Derived from extensive studies on signal transduction pathways in malignant cells, these compounds specifically target the AKT serine/threonine kinase family—consisting of AKT1, AKT2, and AKT3—which plays a central role in regulating cell survival, proliferation, metabolism, and angiogenesis. Dysregulation of AKT signaling, often driven by genetic mutations, amplifications, or aberrant upstream pathway activation, is a hallmark of numerous human cancers, including breast, prostate, colorectal, and glioblastoma. As such, AKT inhibitors have emerged as promising tools to disrupt oncogenic signaling cascades, and their development has been accompanied by intensive scientific inquiry into their molecular mechanisms, pathway interactions, and translational potential.
Akt Inhibitoren/Aktivatoren (Akt Inhibitors/Activators)
| Kat.-Nr. | Produktname | Informationen | Publikationen | Validierung |
|---|---|---|---|---|
| S7863 | SC79 | SC79 ist ein hirngängiger Akt-Phosphorylierungsaktivator und ein Inhibitor der Akt-PH-Domänen-Translokation. |
|

|
| S8019 | AZD5363 (Capivasertib) | Capivasertib (AZD5363) hemmt potent alle Isoformen von Akt (Akt1/Akt2/Akt3) mit IC50 von 3 nM/8 nM/8 nM in zellfreien Assays, ähnlich wie P70S6K/PKA und geringerer Aktivität gegenüber ROCK1/2. Phase 2. |
|

|
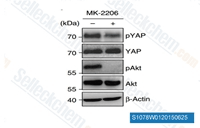
| S1078 | MK-2206 Dihydrochloride | MK-2206 2HCl ist ein hochselektiver Inhibitor von Akt1/2/3 mit einer IC50 von 8 nM/12 nM/65 nM in zellfreien Assays; keine hemmenden Aktivitäten gegen 250 andere Proteinkinasen beobachtet. Diese Verbindung induziert Autophagy und Apoptosis in Krebszellen. Phase 2. |
|
|
| S2808 | Ipatasertib (GDC-0068) | Ipatasertib (GDC-0068, RG7440) ist ein hochselektiver Pan-Akt-Inhibitor, der Akt1/2/3 mit einer IC50 von 5 nM/18 nM/8 nM in zellfreien Assays hemmt und eine 620-fache Selektivität gegenüber PKA aufweist. Diese Verbindung befindet sich derzeit in Phase 2. |
|
-S280804W1120150906.gif)
|
| S1037 | Perifosine | Perifosine ist ein neuartiger Akt-Inhibitor mit einer IC50 von 4,7 M in MM.1S-Zellen, der die Pleckstrin-Homologie-Domäne von Akt angreift. Phase 3. |
|

|
| S1113 | GSK690693 | GSK690693 ist ein pan-Akt-Inhibitor, der Akt1/2/3 mit IC50 von 2 nM/13 nM/9 nM in zellfreien Assays hemmt und auch empfindlich gegenüber der AGC-Kinase-Familie ist: PKA, PrkX und PKC-Isoenzyme. GSK690693 hemmt auch potent AMPK und DAPK3 aus der CAMK-Familie mit IC50 von 50 nM bzw. 81 nM. GSK690693 beeinflusst die Aktivität von Unc-51-like autophagy activating kinase 1 (ULK1) und hemmt robust die STING-abhängige IRF3-Aktivierung. Phase 1. |
|

|
| S1117 | Triciribine (API-2) | Triciribine (API-2) ist ein DNA synthesis Inhibitor, der auch Akt in der PC3-Zelllinie und HIV-1 in CEM-SS-, H9-, H9IIIB-, U1-Zellen mit einer IC50 von 130 nM bzw. 20 nM hemmt; es hemmt nicht PI3K/PDK1 und ist 5000-fach weniger aktiv in Zellen, denen die Adenosin-Kinase fehlt. Phase 1/2. |
|
|
| S7521 | Afuresertib (GSK2110183) | Afuresertib (GSK2110183) ist ein potenter, oral bioverfügbarer Akt-Inhibitor mit Ki-Werten von 0,08 nM, 2 nM und 2,6 nM für Akt1, Akt2 bzw. Akt3 und befindet sich derzeit in Phase-2-Studien. |
|

|
| S2635 | CCT128930 | CCT128930 ist ein potenter, ATP-kompetitiver und selektiver Inhibitor von Akt2 mit einem IC50 von 6 nM in einem zellfreien Assay, 28-fach höhere Selektivität für Akt2 als die eng verwandte PKA-Kinase. Diese Verbindung induziert Zellzyklusarrest, DNA-Schäden und Autophagy unabhängig von der Akt-Inhibition. Eine hohe Dosis dieser Chemikalie löst Zell-Apoptosis in HepG2-Zellen aus. |
|

|
| S2670 | A-674563 HCl | A-674563 HCl ist ein Akt1-Inhibitor mit einem Ki von 11 nM in zellfreien Assays, mäßig potent gegenüber PKA und >30-fach selektiv für Akt1 gegenüber PKC. |
|

|
The PI3K-AKT-MTOR Pathway: A Central Target for AKT Inhibitors
The PI3K-AKT-MTOR pathway is the primary signaling network modulated by AKT inhibitors, serving as a master regulator of cellular homeostasis and malignant transformation. Understanding the architecture and dysregulation of this pathway is fundamental to optimizing the design and application of AKT-targeted therapies.
Pathway Architecture and Physiological Activation
Under physiological conditions, the PI3K-AKT-MTOR pathway is activated by extracellular signals such as growth factors, cytokines, and integrin-mediated cell adhesion. Ligand binding to receptor tyrosine kinases (RTKs) or G protein-coupled receptors (GPCRs) triggers the recruitment and activation of class I PI3K, which phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3)—a lipid second messenger. PIP3 then recruits AKT and phosphoinositide-dependent kinase 1 (PDK1) to the plasma membrane via their pleckstrin homology (PH) domains. PDK1 phosphorylates AKT at threonine 308 (T308), while mammalian target of rapamycin complex 2 (MTORC2) phosphorylates AKT at serine 473 (S473), leading to full activation of AKT. Activated AKT translocates to the cytoplasm and nucleus, where it phosphorylates a diverse array of downstream substrates, orchestrating processes critical for cell survival (e.g., inhibiting BAD and caspase-9), proliferation (e.g., activating cyclin D1), metabolism (e.g., promoting glucose uptake via GLUT4), and angiogenesis (e.g., upregulating VEGF).
Oncogenic Dysregulation of the PI3K-AKT-MTOR Pathway
In cancer cells, the PI3K-AKT-MTOR pathway is frequently dysregulated through multiple mechanisms, driving uncontrolled cell growth and survival. Common alterations include activating mutations in PIK3CA (encoding the p110α catalytic subunit of PI3K), loss-of-function mutations in PTEN (a phosphatase that dephosphorylates PIP3, antagonizing PI3K signaling), amplifications of AKT1/2/3, and overexpression of RTKs (e.g., EGFR, HER2). These aberrations result in constitutive PIP3 accumulation and AKT activation, independent of extracellular signals. For example, PIK3CA mutations are present in ~30% of breast cancers and ~20% of colorectal cancers, leading to hyperactive PI3K signaling and sustained AKT phosphorylation. Similarly, PTEN loss is prevalent in prostate cancer, glioblastoma, and endometrial cancer, removing a key negative regulator of the pathway. Such dysregulation creates a "addiction" of cancer cells to AKT signaling, making them vulnerable to AKT inhibitors.
AKT Inhibitors: Classification, Protein Targeting, and Kinase Selectivity
AKT inhibitors are classified based on their mechanism of action, with distinct classes differing in their ability to target AKT protein conformations, kinase activity, and upstream/downstream signaling components. Achieving selective targeting of AKT kinases while minimizing off-target effects is a key focus of research, as non-specific inhibition can lead to toxicity and reduced therapeutic efficacy.
Classification of AKT Inhibitors by Mechanism
Three main classes of AKT inhibitors have been developed: ATP-competitive inhibitors, allosteric inhibitors, and PH domain inhibitors. ATP-competitive inhibitors bind to the ATP-binding pocket of AKT, preventing ATP hydrolysis and subsequent kinase activity. These inhibitors typically target the active conformation of AKT and may exhibit cross-reactivity with other kinases (e.g., PDK1, SGK family members) due to structural similarities in ATP-binding domains. Examples include capivasertib (AZD5363) and ipatasertib (GDC-0068), which have advanced to late-stage clinical trials for breast and prostate cancer. Allosteric inhibitors, in contrast, bind to a regulatory pocket outside the ATP-binding site, inducing a conformational change that prevents AKT activation. For instance, MK-2206 binds to the PH domain and adjacent regions, blocking AKT recruitment to the plasma membrane and subsequent phosphorylation by PDK1 and MTORC2. Allosteric inhibitors often exhibit higher selectivity for AKT isoforms, reducing off-target kinase inhibition. PH domain inhibitors, a less developed class, directly target the PH domain of AKT, preventing PIP3 binding and membrane localization—an essential step for AKT activation. While promising, these inhibitors face challenges in achieving sufficient potency and bioavailability.
Protein-Protein Interactions and Kinase Selectivity in Inhibitor Design
A major research focus in AKT inhibitor development is optimizing protein targeting and kinase selectivity. AKT isoforms (AKT1, AKT2, AKT3) share high sequence homology in their kinase domains (~80% identity) but exhibit distinct tissue expression patterns and functional roles: AKT1 is widely expressed and critical for cell survival and proliferation; AKT2 is enriched in insulin-responsive tissues (e.g., liver, muscle) and regulates metabolism and cell motility; AKT3 is predominantly expressed in the brain and plays a role in neuronal development. Non-selective AKT inhibitors may disrupt physiological functions of non-oncogenic isoforms, leading to adverse effects such as hyperglycemia (due to AKT2 inhibition) or neurotoxicity (due to AKT3 inhibition). To address this, researchers have pursued isoform-selective inhibitors by leveraging structural differences in the ATP-binding pocket or allosteric sites of AKT isoforms. For example, AKT1-selective inhibitors exploit a unique amino acid residue in the ATP-binding pocket (e.g., alanine at position 230 in AKT1 vs. serine in AKT2/3), enabling selective binding. Additionally, advances in structural biology—including X-ray crystallography and cryo-electron microscopy—have provided detailed insights into AKT protein conformations, facilitating rational drug design.
Functional Impacts of AKT Inhibitors: From Cellular Responses to Preclinical Efficacy
AKT inhibitors exert profound functional effects on cancer cells by disrupting key oncogenic processes, and preclinical research has been instrumental in defining their mechanisms of action, efficacy, and potential resistance pathways. Understanding these functional impacts is critical for translating AKT inhibitors into effective clinical therapies.
Functional Disruption of Oncogenic Processes
AKT inhibitors induce a range of cellular responses in cancer cells, primarily through inhibiting the phosphorylation of AKT substrates. Key functional effects include: (1) Induction of apoptosis: By inhibiting AKT-mediated phosphorylation of BAD (a pro-apoptotic Bcl-2 family member), AKT inhibitors promote BAD dimerization with Bcl-2/Bcl-XL, releasing Bax/Bak to trigger mitochondrial outer membrane permeabilization and caspase activation. (2) Inhibition of cell proliferation: AKT inhibitors block the phosphorylation of p27Kip1 (a cyclin-dependent kinase inhibitor), leading to p27Kip1 accumulation and cell cycle arrest at the G1 phase. They also downregulate cyclin D1 expression, inhibiting progression through the G1-S checkpoint. (3) Suppression of metabolism: AKT inhibitors reduce glucose uptake and glycolysis by inhibiting the translocation of GLUT4 to the plasma membrane and downregulating hexokinase 2 (HK2) and pyruvate kinase M2 (PKM2)—key enzymes in the Warburg effect. (4) Inhibition of angiogenesis: By downregulating VEGF and HIF-1α (hypoxia-inducible factor 1α), AKT inhibitors reduce tumor vascularization, limiting nutrient and oxygen supply to malignant cells. These functional effects are not mutually exclusive; for example, metabolic suppression can enhance apoptotic sensitivity, creating a synergistic anti-tumor response.
Preclinical Efficacy and Combination Therapy Strategies
Preclinical studies using cancer cell lines, patient-derived xenografts (PDXs), and genetically engineered mouse models (GEMMs) have demonstrated the efficacy of AKT inhibitors in multiple cancer types. For instance, capivasertib has shown potent anti-tumor activity in PDX models of triple-negative breast cancer (TNBC) and castration-resistant prostate cancer (CRPC), particularly in tumors with PI3K-AKT pathway alterations. However, monotherapy with AKT inhibitors often leads to the development of resistance, limiting long-term efficacy. Common resistance mechanisms include: (1) Upregulation of alternative survival pathways (e.g., MAPK/ERK, STAT3); (2) Amplification or activation of RTKs (e.g., EGFR, FGFR) that reactivate PI3K-AKT signaling; (3) Mutations in AKT that reduce inhibitor binding (e.g., E17K mutation in AKT1); (4) Activation of MTORC1 via alternative routes (e.g., amino acid signaling). To overcome resistance, researchers have explored combination therapy strategies, such as combining AKT inhibitors with PI3K inhibitors, MTOR inhibitors, RTK inhibitors, or immune checkpoint inhibitors. For example, the combination of capivasertib (AKT inhibitor) and taselisib (PI3K inhibitor) has shown synergistic anti-tumor activity in PIK3CA-mutant breast cancer models, as dual inhibition of PI3K and AKT prevents reactivation of the pathway. Similarly, combining AKT inhibitors with anti-PD-1/PD-L1 antibodies enhances anti-tumor immunity by reducing the expression of immunosuppressive molecules (e.g., PD-L1) on cancer cells and promoting T cell infiltration.
Crosstalk Between AKT Inhibitors and PI3K/MTOR Signaling: Implications for Research and Therapy
The PI3K-AKT-MTOR pathway is characterized by extensive crosstalk and feedback loops, and AKT inhibitors modulate this network in complex ways that impact their therapeutic efficacy. Research into this crosstalk has uncovered critical insights into pathway regulation and identified novel therapeutic opportunities.
PI3K-AKT-MTOR Crosstalk and Feedback Activation
AKT inhibition can trigger feedback activation of upstream components of the PI3K-AKT-MTOR pathway, limiting inhibitor efficacy. For example, AKT phosphorylates and inhibits RTKs (e.g., EGFR) via a negative feedback loop; blocking AKT activity relieves this inhibition, leading to RTK phosphorylation and increased PI3K activation. Similarly, AKT inhibits the adapter protein IRS-1; AKT inhibition enhances IRS-1 stability and PI3K recruitment. Feedback activation can also occur downstream of AKT: AKT phosphorylates and inhibits TSC2 (a negative regulator of MTORC1), so AKT inhibition leads to TSC2 dephosphorylation and reduced MTORC1 activity. However, prolonged MTORC1 inhibition can activate RTK-PI3K signaling via another feedback loop, reactivating AKT. These feedback mechanisms highlight the need for combinatorial targeting of PI3K, AKT, and MTOR to disrupt the pathway comprehensively.
Therapeutic Implications of PI3K/MTOR Co-Targeting
Preclinical and clinical studies have demonstrated that co-targeting PI3K, AKT, and MTOR can overcome feedback activation and improve therapeutic outcomes. For example, combining the PI3K inhibitor alpelisib with the AKT inhibitor capivasertib has shown promising efficacy in PIK3CA-mutant breast cancer, as alpelisib blocks upstream PI3K activation, while capivasertib inhibits AKT, preventing feedback-driven RTK activation. Similarly, combining AKT inhibitors with MTOR inhibitors (e.g., everolimus) targets both AKT and its downstream effector MTORC1, disrupting the pathway at multiple nodes. However, co-targeting can increase toxicity (e.g., hyperglycemia, diarrhea, myelosuppression), so optimizing dosing schedules and patient selection is critical. Biomarker research is also ongoing to identify patients most likely to benefit from PI3K/AKT/MTOR co-targeting, such as those with PIK3CA mutations, PTEN loss, or high AKT phosphorylation levels.
Produkte sind nur für Forschungszwecke bestimmt. Nicht für den menschlichen Gebrauch. Wir verkaufen nicht an Patienten.
©Copyright 2013 Selleck Chemicals. Alle Rechte vorbehalten.